CAPÍTULO 20 Imunodeficiências Congênitas e Adquiridas
CARACTERÍSTICAS GERAIS DAS DOENÇAS DE IMUNODEFICIÊNCIA,
IMUNODEFICIÊNCIAS CONGÊNITAS (PRIMÁRIAS),
IMUNODEFICIÊNCIAS ADQUIRIDAS (SECUNDÁRIAS),
VÍRUS DA IMUNODEFICIÊNCIA HUMANA E SÍNDROME DA IMUNODEFICIÊNCIA ADQUIRIDA,
A integridade do sistema imunológico é essencial para a defesa contra organismos infecciosos e seus produtos tóxicos e, portanto, para a sobrevivência de todos os indivíduos. Os defeitos em um ou mais componentes do sistema imunológico podem levar a distúrbios graves e muitas vezes fatais, que são coletivamente chamados de doenças de imunodeficiência. Estas doenças são classificadas em dois grupos. As imunodeficiências congênitas, ou primárias são defeitos genéticos que resultam no aumento da suscetibilidade à infecção, que é frequentemente manifestado no início da infância e na adolescência, mas às vezes é clinicamente detectado mais tarde na vida. Estima-se que nos Estados Unidos, aproximadamente 1 em cada 500 indivíduos nasce com um defeito em algum componente do sistema imune, embora apenas uma pequena proporção seja afetada de forma grave o suficiente para desenvolver complicações que ameacem a vida. As imunodeficiências adquiridas ou secundárias não são herdadas, mas se desenvolvem como consequência de desnutrição, câncer disseminado, tratamento com fármacos imunossupressores, ou infecção das células do sistema imune, principalmente com o vírus da imunodeficiência humana (HIV), agente etiológico da síndrome da imunodeficiência adquirida (AIDS). Este capítulo descreve os principais tipos de imunodeficiências congênitas e adquiridas, com ênfase em suas patogêneses e nos componentes do sistema imune que estão envolvidos nestes distúrbios.
CARACTERÍSTICAS GERAIS DAS DOENÇAS DE IMUNODEFICIÊNCIA
Antes de começar nossa discussão sobre doenças individuais, é importante resumir algumas características gerais sobre as imunodeficiências.
• A principal consequência da imunodeficiência é um aumento da suscetibilidade à infecção. A natureza da infecção em um determinado paciente depende em grande parte do componente do sistema imune que está com defeito (Tabela 20-1). A imunidade humoral deficiente geralmente resulta no aumento da suscetibilidade à infecção por encapsulados, bactérias de formação de pus e alguns vírus, enquanto os defeitos na imunidade mediada por células levam às infecções por vírus e outros micro-organismos intracelulares. As deficiências combinadas de ambas as imunidades – humoral e mediada por células – tornam os pacientes suscetíveis à infecção por todas as classes de micro-organismos. Os pacientes imunodeficientes geralmente apresentam infecções por micro-organismos que são comumente encontradas, mas efetivamente eliminadas, por pessoas saudáveis; tais infecções são chamadas oportunistas.
• Pacientes com imunodeficiências também são suscetíveis a certos tipos de câncer. Muitos destes cânceres parecem ser provocados por vírus oncogênicos, como o vírus Epstein-Barr. Um aumento da incidência de câncer é visto com mais frequência nas imunodeficiências de células T, pois conforme discutido no Capítulo 17, as células T desempenham um papel importante na vigilância contra vírus oncogênicos e os tumores que eles provocam.
• Paradoxalmente, certas imunodeficiências estão associadas a um aumento na incidência de autoimunidade. O mecanismo subjacente a esta associação não é conhecido.
• A imunodeficiência pode resultar dos defeitos no desenvolvimento ou ativação de linfócitos ou dos defeitos nos mecanismos efetores da imunidade inata e adaptativa. As doenças de imunodeficiência são clínica e patologicamente heterogêneas, em parte porque as doenças diferentes envolvem componentes diferentes do sistema imune.
TABELA 20-1 Características das Imunodeficiências que Afetam os Linfócitos T ou B
| Característica | Deficiência de Células B | Deficiência de Células T |
|---|---|---|
| Suscetibilidade às infecções | Bactérias piogênicas (otite, pneumonia, meningite, osteomielite), bactérias entéricas e vírus, alguns parasitas | Pneumocystis jiroveci, muitos vírus, micobactérias atípicas, fungos |
| Diagnóstico | ||
| Níveis séricos da Ig | Reduzido | Normal ou reduzido |
| Reações DTH para antígenos comuns | Normal | Reduzido |
| Morfologia dos tecidos linfoides | Folículos ausentes ou reduzidos e centros germinativos (zonas das células B) | Geralmente os folículos normais podem ser reduzidos para regiões corticais foliculares (zonas de células T) |
DTH, hipersensibilidade do tipo tardia.
Neste capítulo, primeiro descrevemos as imunodeficiências congênitas, incluindo os defeitos nos componentes do sistema imune inato, e os defeitos nos braços humorais e mediados por células do sistema imune adaptativo. Concluímos com uma discussão sobre imunodeficiências adquiridas, com ênfase sobre a AIDS.
IMUNODEFICIÊNCIAS CONGÊNITAS (PRIMÁRIAS)
Em imunodeficiências congênitas diferentes, a anormalidade etiológica pode estar em componentes do sistema etiológico inato, em estágios diferentes do desenvolvimento dos linfócitos, ou nas respostas de linfócitos maduros à estimulação antigênica. As anormalidades herdadas que afetam a imunidade inata de forma mais comum afetam a via do complemento ou os fagócitos. As anormalidades no desenvolvimento dos linfócitos podem ser provocadas por mutações genéticas que codificam uma variedade de moléculas, incluindo as enzimas, adaptadores, proteínas de transporte e fatores de transcrição. Estes defeitos herdados, e as perturbações orientadas correspondentes nos camundongos, foram úteis para a elucidação dos mecanismos de desenvolvimento dos linfócitos (Cap. 8). As anormalidades no desenvolvimento e na função dos linfócitos B resultam na produção de anticorpos deficientes e são diagnósticos por níveis reduzidos de imunoglobulina (Ig) sérica, respostas de anticorpos defeituosos à vacinação, e, em alguns casos, números reduzidos de células B na circulação ou tecidos linfoides ou células plasmáticas ausentes nos tecidos (Tabela 20-1). As anormalidades na maturação e nas funções dos linfócitos T levam à deficiência de imunidade mediada por células e podem também resultar na produção reduzida de anticorpos. As imunodeficiências de células T primárias são diagnósticas pelos números reduzidos de células T no sangue periférico, baixa reposta proliferativa de linfócitos sanguíneos para ativadores de células T policlonais, tais como a fito-hemaglutinina e reações de hipersensibilidade do tipo tardia (DTH) com deficiência cutânea aos antígenos microbianos ubíquos, tais como os antígenos de Candida. Os defeitos tanto na imunidade humoral como na mediada por células são classificados sob imunodeficiências combinadas graves. Nas seções seguintes, descrevemos as imunodeficiências provocadas por mutações hereditárias nos genes que codificam os componentes do sistema imune inato ou nos genes necessários para o desenvolvimento e ativação de linfócitos. Concluímos com uma breve discussão de estratégias terapêuticas para estas doenças.
Defeitos na Imunidade Inata
A imunidade inata constitui a primeira linha de defesa contra os organismos infecciosos. Dois importantes mediadores da imunidade inata são fagócitos e complementos, ambos os quais também participam nas fases efetoras da imunidade adaptativa. Portanto, os distúrbios congênitos dos fagócitos e o sistema complemento resultam em infecções recorrentes. As deficiências dos complementos foram descritas no Capítulo 12. As deficiências foram descritas nas vias clássicas e alternativas do complemento, bem como na via das lectinas. Eles normalmente apresentam infecções bacterianas recorrentes, principalmente por bactérias encapsuladas e também por espécies de Neisseria, e muitas vezes também contribuem para a suscetibilidade de distúrbios autoimunes, principalmente de lúpus eritematoso sistêmico.
Nesta seção do capítulo, discutimos alguns exemplos de distúrbios de fagócito congênito (Tabela 20-2) e defeitos herdados nas vias do receptor semelhante a Toll (TLR) e na via IL-12/IFN-γ. Os defeitos dos fagócitos geralmente resultam em infecções da pele e no trato respiratório com bactérias ou fungos, este último envolvendo predominantemente espécies de Aspergillus e Candida. Profundos abscessos e estomatites orais também são comuns. Os defeitos na sinalização TLR e na sinalização interferon tipo I podem contribuir com infecções piogênicas recorrentes, bem como com graves infecções virais; os defeitos no IL-12 e na via IFN-γ estão ligados à suscetibilidade aos patógenos intracelulares, particularmente às infecções por micobactérias.
TABELA 20-2 Distúrbios Congênitos da Imunidade Inata
| Doença | Deficiências Funcionais | Mecanismo de Defeito |
|---|---|---|
| Doença granulomatosa crônica | Produção defeituosa de espécies de reativas de oxigênio por fagócitos; infecções bacterianas e fúngicas intracelulares recorrentes | Mutação nos genes do complexo de oxidase dos fagócitos; phox-91 (subunidade a do citocromo b588) é modificada na forma ligada ao X |
| Deficiência de Adesão de Leucócitos tipo 1 | A adesão e a migração de leucócitos defeituosos ligada à redução ou ausência de expressão de integrinas β2; infecções bacterianas e fúngicas recorrentes | As mutações no gene que codificam a cadeia β (CD18) das β2 integrinas |
| Deficiência de adesão de leucócitos tipo 2 | A circulação e a migração de leucócitos defeituosos ligada à redução ou ausência de expressão de ligantes de leucócitos para as E-selectinas e P-selectinas endoteliais, provocando a falha da migração de leucócitos para os tecidos; infecções fúngicas e bacterianas recorrentes. | As mutações no gene que codifica um transportador de GDP-fucose necessário para a síntese do componente sialil Lewis X dos ligantes da E-selectina e P-selectina. |
| Deficiência de adesão de leucócitos tipo 3 | A adesão e migração de leucócitos defeituosos ligadas à sinalização de dentro para fora defeituosa e, portanto, a ativação defeituosa da integrina. | As mutações no gene que codifica o KINDLIN-3 |
| Síndrome Chédiak- Higashi | A fusão das vesículas defeituosas e a função lisossômica nos neutrófilos, macrófagos, células dendríticas, células NK, células T citotóxicas, e muitos outros tipos de células; infecções recorrentes por bactérias piogênicas | A mutação no LYST que leva à exocitose defeituosa dos grânulos de secreção e à função lisossômica |
| Defeitos de sinalização do receptor semelhante a Toll | As infecções recorrentes devido aos defeitos na sinalização do TLR e do CD40 e a produção defeituosa de interferon do tipo I | As mutações no NEMO, UNC93B, MyD88, IκBα e no IRAK-4 comprometem a ativação de NF-κB dos receptores semelhantes a Toll |
IRAK-4, a cinase 4 associada ao receptor IL-1; LYST, proteína de tráfico lisossômico; NEMO, modulador essencial NF-κB.
Atividades Microbicidas Defeituosas dos Fagócitos: Doença Granulomatosa Crônica
A doença granulomatosa crônica (CGD) é provocada por mutações nos componentes do complexo enzimático da oxidase fagocitária (phox). É uma doença rara, estimada afetar cerca de 1 em 1 milhão de indivíduos nos Estados Unidos. Cerca de dois terços de casos mostram um padrão de herança recessivo ligado ao X, e o restante é autossômico recessivo. A forma de doença mais comum ligada ao X é provocada por uma mutação no gene que codifica a subunidade 91 kD α do citocromo b558, uma proteína da membrana integral, também conhecida como phox-91. Esta mutação resulta na produção defeituosa do ânion superóxido, uma das várias espécies reativas de oxigênio, que constituem um grande mecanismo microbicida dos fagócitos (Cap. 4). As mutações em outros componentes do complexo phox contribuem para as variantes recessivas autossômicas do CGD. A produção defeituosa das espécies reativa de oxigênio resulta em falha para matar micro-organismos fagocitados. A doença é caracterizada por infecções recorrentes com bactérias e fungos intracelulares produtores de catalase, geralmente desde a primeira infância. Muitos dos organismos que são particularmente problemáticos nos pacientes CGD produzem catalase, que destrói o peróxido de hidrogênio microbicida que pode ser produzido por células hospedeiras do radical superóxido do oxigênio reativo residual. Como as infecções não são controladas por fagócitos, elas estimulam respostas imunes mediadas por células crônicas, resultando na ativação de macrófagos mediados por células T e na formação de granulomas compostos por macrófagos ativados. Presumivelmente, estes macrófagos ativados tentam limitar ou eliminar os micro-organismos apesar da produção defeituosa de espécies reativa de oxigênio. Esta aparência histológica é a base para o nome do distúrbio. A doença é frequentemente fatal, mesmo com a terapia agressiva à base de antibióticos.
A citocina interferon-γ (IFN-γ) aumenta a transcrição do gene que codifica o phox-91 e também estimula outros componentes do complexo enzimático da oxidase fagocitária. Portanto, o IFN-γ estimula a produção de superóxido através de neutrófilos normais, assim como por neutrófilos CGD, especialmente nos casos em que a porção codificada do gene phox-01 está intacta, mas sua transcrição está reduzida. Uma vez que a produção de superóxido é restaurada para cerca de 10% dos níveis normais, a resistência à infecção é bastante aprimorada. A terapia IFN-γ é agora comumente usada para o tratamento de CGD ligado a X.
Deficiências de Adesão de Leucócitos
As deficiências de adesão de leucócitos são um grupo de distúrbios autossômicos recessivos provocados por defeitos nas moléculas de adesão leucocitárias e endoteliais. Estas doenças são caracterizadas por uma falha nos leucócitos, principalmente nos neutrófilos, o recrutamento para os locais de infecção, resultando em periodontite grave e outras infecções recorrentes no início da vida, e a incapacidade de formar pus. Tipos diferentes de deficiências de adesão de leucócitos são provocados por mutações em genes diferentes.
• A deficiência de adesão de leucócitos tipo 1 (LAD-1) é um distúrbio recessivo autossômico raro caracterizado por infecções bacterianas e fúngicas recorrentes e cicatrização de ferimentos prejudicada. Nestes pacientes, a maioria das funções dependentes de adesão é anormal. Estas funções incluem a adesão ao endotélio, agregação dos neutrófilos e quimiotaxia, fagocitose e citotoxicidade mediada por neutrófilos, células NK e linfócitos T. A base molecular do defeito é a expressão ausente ou deficiente das integrinas β2 (heterodímeros do CD18 e a família CD11 das glicoproteínas), devido a várias mutações no gene CD18. As β2 integrinas incluem os antígenos associados às funções leucocitárias 1 (LFA-1 ou CD11aCD18), Mac-1 (CD11bCD18) e p150,95 (CD11cCD18). Estas proteínas participam na adesão dos leucócitos a outras células, nomeadamente células endoteliais, e a ligação dos linfócitos T com células apresentadoras de antígenos (APC) (Cap. 3).
• A deficiência de adesão de leucócitos do tipo 2 (LAD-2) é outro distúrbio raro que é clinicamente semelhante ao LAD-1, mas não é devido aos defeitos da integrina. Em contraste, o LAD-2 resulta da ausência de sialil Lewis X, o ligante do carboidrato tetrassacarídeo nos neutrófilos e em outros leucócitos, que é necessário para a ligação com a E-selectina e a P-selectina no endotélio ativado por citocinas (Cap. 3). Este defeito é provocado por uma mutação em um transportador de GDP-fucose responsável pelo transporte de fucose no Golgi, resultando na incapacidade de sintetizar sialil Lewis X. A ausência de resultados de sialil Lewis X na ligação defeituosa de leucócitos com o endotélio, a ausência de leucócitos “rolando”, e, portanto o recrutamento defeituoso de leucócitos para os locais de infecção. Esta anormalidade na fucosilação vista no LAD-2 também contribui para um fenótipo de grupo sanguíneo Bombaim provocado pela ausência de todos os antígenos de grupos sanguíneos ABO, bem como para o retardo mental e outros defeitos referentes ao desenvolvimento. A fucose é um componente essencial do glicolipídeo H que forma o antígeno principal no sistema ABO.
• A deficiência de adesão de leucócitos tipo 3 (LAD-3) envolve um defeito na sinalização de dentro para fora e, assim, um defeito na ativação da integrina induzida pela quimiocina, que é necessária para os leucócitos se ligarem firmemente ao endotélio (Cap. 3). Em um subgrupo de pacientes, ela é provocada por mutações no gene que codificam a KINDLIN-3. A KINDLIN-3 é uma proteína que se liga à cauda citoplasmática de algumas integrinas e está envolvida na sinalização. O aumento do sangramento também é observado em indivíduos com mutações de KINDLIN-3 devido à disfunção da integrina nas plaquetas.
Defeitos nas Células NK e em Outros Leucócitos: A Síndrome de Chédiak-Higashi
A síndrome de Chédiak-Higashi é um distúrbio recessivo autossômico raro caracterizado por infecções recorrentes por bactérias piogênicas, albinismo oculocutâneo parcial e infiltração de diversos órgãos por linfócitos não neoplásticos. Os neutrófilos, monócitos e linfócitos destes pacientes contêm lisossomos gigantes. Esta doença é provocada por mutações no gene que codifica a proteína reguladora do tráfico lisossômico LYST, resultando na fusão do lisossomo com o fagossomo defeituosa nos neutrófilos e macrófagos (provocando resistência reduzida à infecção), formação defeituosa de melanossoma nos melanócitos (provocando o albinismo), e anormalidades lisossômicas nas células do sistema nervoso (provocando defeitos nos nervos) e plaquetas (levando a distúrbios hemorrágicos). Os lisossomos gigantes se formam nos neutrófilos durante a maturação destas células de precursores mieloides. Alguns destes precursores de neutrófilos morrem prematuramente, resultando em leucopenia moderada. Os neutrófilos sobreviventes podem conter níveis reduzidos de enzimas lisossômicas que normalmente funcionam na morte microbiana. Estas células também são defeituosas na quimiotaxia e na fagocitose, também contribuindo para sua atividade microbicida deficiente. A função das células NK nestes pacientes é prejudicada, provavelmente devido a uma anormalidade nos grânulos citoplasmáticos que armazenam as proteínas que medeiam a citotoxicidade. A gravidade do defeito na função dos linfócitos T citotóxicos (CTL) é variável entre os pacientes. Uma cepa do rato mutante chamado rato bege é um modelo de animal para a síndrome de Chédiak-Higashi. Esta espécie é caracterizada pela função das células NK deficientes e lisossomos gigantes nos leucócitos. A mutação bege foi mapeada para o lócus do rato Lyst.
Outras mutações que afetam tanto a função das células NK como das células CTL serão consideradas posteriormente, quando discutirmos os defeitos na ativação e função do linfócito T. Uma mutação no CD16/FcγRIII, o receptor Fc nas células NK, que é necessário para a citotoxicidade celular dependente de anticorpos (Cap. 12), foi descrita em um paciente com infecções virais recorrentes.
Defeitos Herdados nas Vias TLR, Sinalização do Fator Nuclear κB e Interferons do Tipo I
Os defeitos hereditários nas respostas dependentes de TLR são raros e foram reconhecidos apenas recentemente. A principal sinalização via abaixo da maioria dos TLRs, bem como do receptor da interceucina 1 (IL-1R) envolve o adaptador MyD88 e as cinases IRAK-4 e IRAK-1 (Cap. 4), e esta via resulta no fator nuclear κB (NF-κB) – indução dependente das citocinas pró-inflamatórias. O TLR 3, 7, 8 e o 9 que reconhecem ácidos nucleicos, estão localizados nos endossomos e requerem uma proteína chamada UNC93B para sua função. A UNC93B é uma proteína da membrana do retículo endoplasmático que interage com os TLR endossômicos quando eles são sintetizados no retículo endoplasmático e ajudam a entregar esses TLR aos endossomos. A proteína UNC93B também é crítica para a sinalização de TLR específicos do ácido nucleico. Sinalização nos TLR endossomais resulta na síntese e secreção dos interferons do tipo I. Os defeitos na sinalização TLR tendem a ter um fenótipo clínico bastante circunscrito. As infecções bacterianas invasivas graves no início da vida, principalmente doenças pneumocócicas, são observadas nos indivíduos com mutações no MYD88 e no IRAK4. Posteriormente na vida, as infecções tendem a ser menos graves. As mutações heterozigóticas no TLR3, assim como as mutações homozigóticas no UNC93B resultam na geração reduzida de interferon tipo I e na suscetibilidade à encefalite por herpes simples. Os receptores de interferon tipo I ativam o fator de transcrição STAT1. As mutações de perda de função STAT1 (que interferem na sinalização do interferon) também foram associadas às infecções virais graves, nomeadamente encefalite por herpes simples.
Algumas deficiências imunológicas são provocadas por defeitos nas vias de sinalização de TLR. As mutações pontuais no inibidor da cinase κB γ (IKKγ), também conhecidas como moduladores essenciais dos fatores nucleares κB (NEMO), um componente do complexo da cinase IκB que é necessário para a ativação de NF-κB, contribui para a condição recessiva ligada ao X, conhecida como displasia ectodérmica anidrótica com imunodeficiência (EDA-ID). Neste distúrbio, a diferenciação das estruturas derivadas do ectoderma é anormal, e a função imunológica é prejudicada de diversas maneiras. As respostas aos sinais TLR, bem como aos sinais CD40 ficam comprometidas. Estes pacientes sofrem de infecções com bactérias piogênicas encapsuladas, assim como com patógenos bacterianos intracelulares incluindo micobactérias, vírus e fungos, como o Pneumocystis jiroveci (veja também discussão posterior na seção das síndromes de hiper-IgM). Uma forma recessiva autossômica do EDA-ID foi descrita em que uma mutação pontual hipermórfica no IκBα impede a fosforilação, ubiquitinação e degradação do IκBα, levando assim à ativação prejudicada do NF-κB.
Defeitos na Via IL-12/IFN-γ
O IL-12 é secretado pelas células dendríticas e macrófagos, e a sinalização do IL-12R induz à síntese de IFN-γ pelas células T auxiliares (helper), células T citotóxicas e células NK (Cap. 4). As mutações nos genes que codificam a IL-12p40, a cadeia IL-12Rβ1, e ambas as cadeias do receptor IFN-γ, bem como de algumas mutações hipomórficas no STAT1, todas resultam na suscetibilidade às espécies de Micobactérias ambientais (muitas vezes chamadas de micobactérias atípicas), tais como as Mycobacterium avium, Mycobacterium kansasii e as Mycobacterium fortuitum. As mutações IKKγ/NEMO também levam à suscetibilidade a patógenos intracelulares, incluindo as micobactérias, conforme discutido na seção anterior.
Imunodeficiências Combinadas Graves
As imunodeficiências que afetam tanto a imunidade humoral quanto a mediada por células são chamadas imunodeficiências combinadas, e um subgrupo destas em que a maioria de células T periféricas está faltando ou com defeito é conhecido como imunodeficiências combinadas graves (SCID) (Tabela 20-3). Estas doenças são caracterizadas por deficiências de ambas as células B e T ou somente das células T; em último caso, o defeito na imunidade humoral se deve à ausência de célula T auxiliar. As crianças com SCID costumam ter infecções durante seu primeiro ano de vida, a pneumonia Pneumocystis jiroveci é particularmente comum, e elas sucumbem a estas infecções, a menos que sejam tratadas.
TABELA 20-3 Imunodeficiências Combinadas Graves
| Doença | Deficiências Funcionais | Mecanismo de Defeito |
|---|---|---|
| Defeitos na sinalização de citocinas | ||
| SCID ligada ao X | A redução acentuada nas células T; células B normais ou aumentadas; Ig sérica reduzida | As mutações da cadeia comum γ do receptor da citocina; desenvolvimento defeituoso das células T na ausência de sinais derivados de IL-7 |
| Formas recessivas autossômicas | Redução acentuada nas células T; células B normais ou aumentadas; Ig sérica reduzida | Mutações nos IL2RA, IL7RA, JAK3 |
| Defeitos nas vias de salvamento dos nucleotídeos | ||
| Deficiência de ADA | Redução progressiva nas células T, B e NK; Ig sérica reduzida | A deficiência de ADA provocada pelas mutações no gene, levando ao acúmulo de metabólitos tóxicos nos linfócitos |
| Deficiência de PNP | Redução progressiva nas células T, B e NK; Ig sérica reduzida | A deficiência de PNP provocada pelas mutações no gene, levando ao acúmulo de metabólitos tóxicos nos linfócitos |
| Defeitos na recombinação de V(D)J | ||
| Recombinação da deficiência de RAG1 ou RAG2* | Redução das células T e B; Ig sérica reduzida; ausência ou deficiência de células T e B | Defeito de clivagem durante a recombinação de V(D)J; mutações no RAG1 ou RAG2 |
| Reparo da quebra da fita dupla e ponto de controle | Redução das células T e B; Ig sérica reduzida; ausência ou deficiência de células T e B | Falha em resolver os ganchos durante a recombinação de V(D)J; mutações no ARTEMIS, DNA-PKcs, CERNUNNOS, LIG4, NBS1, MRE11, ATM |
| Desenvolvimento de timo defeituoso | ||
| Controle do ponto pré-TCR defeituoso | Redução de células T; células B normais ou reduzidas; Ig sérica reduzida | Mutações no CD45, CD3D, CD3E, ORAI1 (Componente do canal CRAC), STIM1 |
| Síndrome de DiGeorge | Redução de células T; células B normais; Ig sérica normal ou reduzida | Exclusão 22ql 1; T-box 1 (TBX1) mutações do fator de transcrição |
| Deficiência de FoxN1 | Aplasia do timo com o desenvolvimento das células defeituosas do timo | Mutação recessiva no FOXN1 |
| Outros defeitos | ||
| Disgenesia reticular | Redução de células T, B e mieloides | Mutação no AK2 |
ADA, adenosina deaminase; AK2, adenilato cinase 2; ATM, ataxia-telangiectasia modificada; CRAC, canal ativado de liberação de cálcio; DNA-PKcs, subunidade catalítica da proteína cinase dependente de DNA; LIG4, DNA ligase 4; MRE11, recombinação meiótica homóloga 11; NBS1, Síndrome de interrupção de Nijmegen 1; PNP, purina nucleosídeo fosforilase.
* As mutações hipomórficas nos RAG genes e no ARTEMIS podem contribuir para a síndrome de Omenn.
O SCID resulta do desenvolvimento de linfócitos T prejudicados com ou sem defeitos na maturação das células B (Fig. 20-1). O epitélio do timo contribui de forma decisiva para o desenvolvimento inicial de células T. O processo de maturação dos linfócitos T (e B) de células-tronco hematopoiéticas para linfócitos maduros competentes funcionalmente envolve a proliferação de progenitores de linfócitos no início, rearranjo do lócus que codifica uma cadeia do receptor do antígeno seguida pela seleção das células que se formaram nos rearranjos produtivos dentro da estrutura em um ponto de conferência do receptor pré-antígeno, expressão de ambas as cadeias do receptor do antígeno, e seleção das células com especificidades úteis (Cap. 8). Defeitos em muitos destes passos foram descritos em diferentes formas de SCID. Cerca de 50% dos SCID são recessivos autossômicos; o resto é ligado ao X. A causa mais comum do SCID recessivo autossômico é a deficiência da enzima deaminase adenosina, necessária para o metabolismo das purinas. O SCID ligado ao X é provocado por mutações no gene que codificam um componente do receptor de citocina chamado cadeia γ comum. Os distúrbios individuais são descritos aqui.
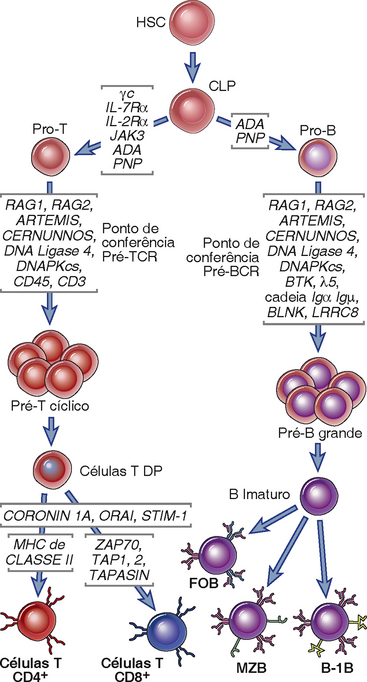
FIGURA 20-1 Imunodeficiência provocada por defeitos na maturação das células B e T. São mostradas as imunodeficiências primárias provocadas por defeitos genéticos na maturação dos linfócitos. Estes defeitos podem afetar a maturação isolada da célula T, a maturação isolada da célula B, ou de ambas. CLP, progenitor linfoide comum; DP, duplo positivo; FoB, células B foliculares; HSC, células-tronco hematopoiéticas; MZB, células B de zona marginal.
A Síndrome de DiGeorge e Outras Formas de SCID, devido ao Defeito do Desenvolvimento Epitelial Tímico
A falha ou desenvolvimento incompleto do primórdio do timo pode levar a um desenvolvimento defeituoso da célula T. O defeito mais comum no desenvolvimento tímico ligado ao SCID é viso em crianças com síndrome de DiGeorge. Esta deficiência de células T seletivas se deve a uma malformação congênita que resulta no desenvolvimento defeituoso do timo e das glândulas paratireoides, bem como de outras estruturas que se desenvolvem da terceira e quarta bolsas faríngeas durante a vida fetal. O defeito congênito é manifesto pela hipoplasia ou agenesia do timo, que leva à deficiência de maturação das células T, ausência de glândulas paratireoides provocando homeostase do cálcio anormal e tremor muscular (tetania), desenvolvimento anormal dos grandes vasos e deformidades faciais. Os pacientes diferentes podem apresentar graus variáveis destas anormalidades. A doença é provocada mais frequentemente por uma supressão no cromossomo 22q11. As mutações no homólogo murino de um gene que codifica um fator de transcrição chamado T-box 1 (TBX1), que fica dentro da região suprimida na síndrome de DiGeorge, também resultam em um defeito semelhante no desenvolvimento do timo. É provável que a imunodeficiência associada à síndrome de DiGeorge possa ser explicada, pelo menos em parte, pela supressão do gene TBX1. Nesta síndrome, os linfócitos T do sangue periférico estão ausentes ou tem sua quantidade muito reduzida, e as células não respondem aos ativadores de células T policlonais ou nas misturas de leucócitos misturados. Os níveis de anticorpos geralmente são normais, mas podem ser reduzidos em pacientes gravemente afetados. Como em outras deficiências graves de células T, os pacientes estão suscetíveis à micobactérias, infecções virais e fúngicas.
A imunodeficiência associada à síndrome de DiGeorge pode ser corrigida através do transplante fetal de timo ou através do transplante de medula óssea. Esse tratamento normalmente não é necessário, no entanto, porque a função das células T tende a melhorar com a idade em uma grande fração de pacientes com esta síndrome geralmente fica normal em 5 anos. A melhora com a idade provavelmente ocorre devido à presença de algum tecido tímico ou porque alguns locais extratímicos ainda não definidos assumem a função da maturação das células T. Também é possível que, à medida que estes pacientes ficam mais velhos, o tecido do timo se desenvolva em locais ectópicos (ou seja, em locais atípicos).
Um modelo animal de imunodeficiência em célula T resultante do desenvolvimento anormal do timo é o rato nude (atímico). Estes ratos têm um defeito hereditário de determinados tipos de células epiteliais na pele, levando à ausência de pelos e no revestimento da terceira e quarta bolsas da faringe, provocando hipoplasia tímica. O transtorno é devido a uma mutação no gene FoxN1 que codifica um fator de transcrição da família Forkhead que é necessário para o desenvolvimento normal de determinados tipos de células derivadas do ectoderma. Os ratos afetados têm timos rudimentares, nos quais a maturação das células T não pode ocorrer normalmente. Como resultado, poucas ou nenhuma célula T madura está presente nos tecidos linfoides periféricos, e as reações imunes mediadas pelas células não podem ocorrer. As mutações do FOXN1 recessivo autossômico têm sido descritas em um pequeno número de pacientes que se apresentam com o SCID, alopecia (queda de cabelo) e distrofia ungueal. Um defeito ainda mais raro no timo foi descrito envolvendo uma mutação no CORONIN-1A, que codifica uma proteína que regula o citoesqueleto da actina. A ausência do CORONIN-1A funcional resulta na saída dos defeitos das células T maduras do timo.
Deficiência de ADA e Outras Formas de SCID Provocadas pelos Defeitos no Metabolismo dos Nucleotídeos
A causa mais comum do SCID recessivo autossômico é a deficiência de uma enzima chamada adenosina deaminase (ADA), devido às mutações no gene da ADA. As funções da ADA na via de salvamento da síntese da purina e catalisa a desaminação da adenosina irreversível da adenosina e 2′-deoxiadenosina para inosina e 2′-deoxinosina, respectivamente. A deficiência da enzima leva ao acúmulo de deoxiadenosina e seus precursores S-adenosil homocisteína e deoxiadenosina trifosfato (dATP). Estes subprodutos têm muitos efeitos tóxicos, incluindo a inibição da síntese de DNA. Embora a ADA esteja presente na maioria das células, o desenvolvimento de linfócitos é menos eficiente do que na maioria de outros tipos de células na degradação da dATP em 2′-deoxiadenosina, e, portanto, a maturação dos linfócitos é particularmente sensível à deficiência de ADA. Outras características da doença podem incluir surdez, anomalias costocondrais, lesões no fígado e problemas comportamentais. A deficiência de ADA leva à redução do número de células B e T; os números de linfócitos são geralmente normais no nascimento, mas caem vertiginosamente durante o primeiro ano de vida. Alguns pacientes podem ter um número praticamente normal de células T, mas estas células não proliferam na resposta à estimulação antigênica.
Uma forma recessiva autossômica mais rara de SCID é devida à deficiência da purina nucleosídeo fosforilase (PNF), uma enzima que também está envolvida no catabolismo da purina. A PNF catalisa a conversão da inosina para hipoxantina e da guanosina para guanina, e a deficiência de PNF leva ao acúmulo de deoxiguanosina e deoxiguanosina trifosfato, com efeitos tóxicos nos linfócitos imaturos, principalmente nas células T. A anemia hemolítica autoimune e a deterioração neurológica progressiva também são características deste distúrbio.
Uma forma particularmente grave de SCID é vista em uma doença chamada disgenesia reticular. Esta doença rara é caracterizada pela ausência dos linfócitos T e B e a maioria das células mieloides, incluindo os granulócitos, e é devido a um defeito no desenvolvimento dos progenitores dos linfoides e dos mieloides. Esta doença recessiva autossômica se deve a uma mutação no gene adenilato cinase 2 (AK2). A proteína AK2 regula o nível de adenosina difosfato, e na ausência de AK2 há aumento de apoptose de precursores linfoides e mieloides.
SCID Ligado ao X
O SCID ligado ao X é provocado por mutações no gene que codifica a cadeia comum γ (γc) compartilhada pelos receptores para as interleucinas IL-2, IL-4, IL-7, IL-9 e IL-15 (Caps. 4 e 9). O SCID ligado ao X é caracterizado pela deficiência na maturação de células T e células NK e números muito reduzidos de células maduras T e NK, mas o número de células B é geralmente normal ou aumentado. A imunodeficiência humoral nesta doença se deve a uma falta de auxílio das células T para a produção de anticorpos. Esta doença é resultado da incapacidade da citocina linfopoiética IL-7, cujo receptor utiliza a cadeia γc para sinalização, para estimular o crescimento dos timócitos imaturos. Além disso, o receptor para o IL-15, que é um forte estímulo para a proliferação de células NK, também usa a cadeia de sinalização γc, e a falta da função IL-15 é responsável pela deficiência de células NK.
As mulheres heterozigotas são geralmente portadoras fenotipicamente normais, enquanto os homens que herdam o cromossomo anormal X manifestam a doença. Como as células de desenvolvimento nas mulheres inativam aleatoriamente um dos dois cromossomos X, o alelo normal que codifica uma proteína funcional γc não será expressa na metade dos precursores dos linfócitos em uma mulher portadora. Estas células não conseguirão amadurecer e, consequentemente, todos os linfócitos maduros em uma mulher portadora terão inativado o mesmo cromossomo X (portadores do alelo mutante). Em contraste, metade de todas as células não linfoides terá um cromossomo X inativado, e metade do outro. A comparação da inativação do cromossomo X nas células linfoides versus as células não linfoides pode ser usada para identificar portadores de alelos mutantes. O uso não aleatório dos cromossomos X nos linfócitos maduros também é característico de mulheres portadoras de outras mutações de genes ligados ao X que afetam o desenvolvimento dos linfócitos, conforme discutido posteriormente.
Mutações Recessivas Autossômicas nos Componentes de Sinalização das Citocinas
Alguns pacientes com uma doença idêntica ao SCID ligado ao X mostram uma herança recessiva autossômica. Estes pacientes têm mutações no receptor IL-7 da cadeia α ou a JAK3 cinase, que se associa à cadeia γc e é necessária para a sinalização por este receptor (Cap. 7). Os pacientes com mutações no gene que codifica o IL-7R da cadeia α têm um defeito no desenvolvimento das células T, mas apresentam desenvolvimento normal das células NK, porque a sinalização do IL-15 não é afetada, e têm números normais de células B.
Imunodeficiência Combinada Grave Provocada pelos Defeitos na Recombinação do V(D)J e Sinalização do Ponto de Controle do Pré-TCR
A ausência da recombinação de V(D)J leva a uma incapacidade de expressar o pré-TCR e o pré-BCR e um bloco de desenvolvimento de células T e B. As mutações nos genes RAG1 ou RAG2 (cujos produtos das proteínas medeiam o passo da clivagem durante a recombinação do V(D)J) ou o gene ARTEMIS, que codifica uma endonuclease que resolve os grampos de codificação final durante a recombinação de V(D)J, todos resultam no fracasso da recombinação do V(D)J. Estas doenças são raras, mas elas são responsáveis por um grande número de formas recessivas autossômicas de SCID. As funções normais destes genes são discutidas no Capítulo 8. Nas crianças com estas mutações, os linfócitos B e T estão ausentes e a imunidade está seriamente comprometida. As mutações nos genes que codificam as proteínas envolvidas na junção final de reparo/não homóloga de quebra de cadeia dupla de DNA também levam ao SCID devido aos defeitos na recombinação de V(D)J. As mutações homozigóticas no gene que codifica a subunidade catalítica da proteína cinase dependente de DNA (DNA-PK), CERNUNNOS/XLF e DNA LIGASE 4; todos levam ao SCID. Entre as muitas funções da DNA-PK estão a fosforilação e ativação da ARTEMIS, e o CERNUNNOS interage com o complexo XRCC4/DNA ligase 4 e, presumivelmente, facilita o evento de ligação que completa o processo de junção final não homóloga. Os defeitos genéticos neste processo de junção final também resultam no aumento da sensibilidade celular à radiação e podem resultar em outras manifestações, tais como microcefalia, dismorfismos faciais e desenvolvimento de dente defeituoso.
As mutações hipomórficas (que reduz a função apenas parcialmente) nos genes RAG, no ARTEMIS, ou no gene IL7RA são a causa de um distúrbio caracterizado pela geração restrita de células T e B, imunodeficiência e desregulação da imunidade. Este distúrbio é conhecido como síndrome de Omenn. Ela é fenotipicamente diferente dos distúrbios descritos porque nesta doença a imunodeficiência coexiste com a ativação imune exagerada e a autoimunidade. Isto pode ser provocado pela ausência relativa das células T reguladoras, ou em casos com redução da recombinação V(D)J, edição do receptor defeituoso nas células B imaturas.
Embora a maioria das formas recessivas autossômicas de SCID seja ligada às mutações na ADA, RAG1, RAG2 e ARTEMIS, as formas raras desta síndrome são provocadas pelas mutações nos genes que codificam a fosfatase CD45 (que é um regulador positivo da família das cinases Src, tais como Fyn, Lck e Lyn) e as mutações no CD3 δ ou cadeias ε ou na cadeia ζ associada ao CD3. Estas mutações contribuem para a sinalização pré-TCR defeituosa e resultam em um bloqueio no desenvolvimento de células Tαβ.
A Síndrome dos Linfócitos Essenciais e Outros Defeitos na Seleção Positiva de Células T
A geração de células T CD4+ e CD8+ positivas de timócitos positivos depende da seleção positiva e de eventos de compromisso com a linhagem. As mutações hereditárias específicas nos genes que regulam o processo da seleção positiva revogam o desenvolvimento das células T CD4+ ou das células T CD8+.
A deficiência do complexo principal de histocompatibilidade (MHC) de classe II, também chamada de síndrome do linfócito essencial, é um grupo heterogêneo raro de doenças recessivas autossômicas em que os pacientes expressam pouco ou nenhum HLA-DP, HLA-DQ, ou HLA-DR nos linfócitos B, macrófagos, e células dendríticas e falham ao expressar as moléculas MHC de classe II em resposta ao IFN-γ. Eles expressam níveis normais ou apenas ligeiramente reduzidos de moléculas MCH de classe I e β2-microglobulina. A maioria dos casos de síndrome de linfócitos essenciais se deve às mutações nos genes que codificam as proteínas que regulam a transcrição dos genes de MHC de classe I. Por exemplo, as mutações que afetam o fator de transcrição expresso constitutivamente RFX5 ou o ativador transcricional induzível IFN-γ CIITA levam à expressão reduzida do MHC de classe II MHC e à falha dos APCs em ativar os linfócitos T CD4+. A falha da apresentação do antígeno pode resultar na seleção positiva defeituosa das células T no timo, com a redução do número de células maduras T CD4+ ou ativação de células defeituosas na periferia. Os indivíduos afetados são deficientes nas respostas ao DTH e nas respostas dos anticorpos aos antígenos proteicos dependentes das células T. A doença aparece no primeiro ano de vida e é geralmente fatal a menos que seja tratada com transplante de medula óssea.
As deficiências do MHC de classe I recessivas autossômicas também foram descritas e são caracterizadas pela diminuição do número e função de células T CD8+. Em alguns casos, a incapacidade de expressar as moléculas MHC de classe I se deve às mutações nos genes TAP-1 ou TAP-2, que codificam as subunidades do complexo do TAP (transportador associado ao processamento de antígeno), que normalmente transporta peptídeos do citosol para o retículo endoplasmático, onde eles são necessários para a montagem do MHC de classe I (Cap. 6). Estes pacientes com deficiência em TAP expressam poucas moléculas MHC de classe I de superfície celular, um fenótipo semelhante ao gene TAP dos camundongos eliminados. Estes pacientes sofrem principalmente de lesões granulomatosas necrotizantes na pele e infecções bacterianas no trato respiratório, mas não infecções virais, o que é surpreendente considerando que a função principal das células T CD8+ é a defesa contra os vírus. Uma deficiência semelhante da expressão MHC da classe I foi observada nos pacientes com mutações no gene que codifica a proteína tapasina (Cap. 6).
Os pacientes com deficiência de ZAP-70 têm um defeito de comprometimento da linhagem, resultando nas células T CD8+ reduzidas, mas não nas células T CD4+; a razão para a perda seletiva não é clara. Este defeito específico da tirosina cinase não compromete o desenvolvimento das células T CD4+ ou a emigração para a periferia. No entanto, estas células T CD4+ não conseguem proliferar normalmente quando desafios com antígenos.
SCID Provocado pela Ativação de Células T Defeituosas
Outra forma rara de SCID é provocada pela mutação em um gene que codifica Orai1, um componente do canal CRAC (Cap. 7). A sinalização do receptor do antígeno leva à ativação da isoforma γ da fosfolipase C (PLCγ) e a liberação dependente de inositol trifosfato (IP3) de íons de cálcio a partir do retículo endoplasmático e da mitocôndria (Cap. 7). O cálcio liberado é reabastecido por um estoque operado por canais de CRAC que facilitam um influxo de cálcio extracelular. Este processo é crucial para a ativação do linfócito, e é defeituoso nas células com ORAI1 mutante. Um fenótipo semelhante é observado nos pacientes com mutações no STIM1, que codifica uma proteína do retículo endoplasmático que detecta a depleção dos depósitos de cálcio e contribui para a abertura do canal CRAC. Pacientes com mutações no ORAI1 e STIM1 não apresentam um defeito no desenvolvimento das células T, mas suas células T não podem ser adequadamente ativadas.
Deficiências de Anticorpos: Defeitos no Desenvolvimento e Ativação das Células B
Enquanto os defeitos no desenvolvimento das células T ou em ambos os desenvolvimentos das células T e B contribuem para o fenótipo SCID, mais defeitos circunscritos nas células B resultam em distúrbios em que a anormalidade primária está na síntese dos anticorpos (Tabela 20-4). Algumas destas doenças são provocadas pelos defeitos no desenvolvimento das células B (Fig. 20-1) e outras pela ativação anormal de células B e produção de anticorpos (Fig. 20-2). No entanto, em um subgrupo de síndromes hiper-IgM discutidas posteriormente, as deficiências de anticorpos também são acompanhadas por defeitos na ativação de macrófagos e APC, que por sua vez resultam na imunidade mediada por células atenuadas.
TABELA 20-4 Deficiências de Anticorpos
| Doença | Deficiências Funcionais | Mecanismo de Defeito |
|---|---|---|
| Agamaglobulinemias | ||
| Ligadas ao X | Redução de todos os isótipos da Ig sérica; número de células B reduzido | Defeito no ponto de controle do receptor de pré-B; mutação Btk |
| Formas recessivas autossômicas | Redução de todos os isótipos da Ig sérica; número de células B reduzido | Defeito no ponto de controle do receptor do pré-B; mutações na cadeia pesada IgM (), cadeias leves substitutas (λ5), Igα, BLNK |
| Hipogamaglobulinemias/defeitos do isótipo | ||
| Deficiência seletiva de IgA | Redução de IgA; pode estar associada ao aumento da suscetibilidade às infecções bacterianas e a protozoários como o Giardia lamblia | As mutações no TACI em alguns pacientes |
| Deficiência seletiva de IgG2 | Aumento da suscetibilidade às infecções bacterianas | Um pequeno subgrupo tem exclusão no lócus IgH γ2 |
| Imunodeficiência variável comum | Hipogamaglobulinemia; quantidade de células B normal ou reduzida | Mutações no ICOS e TACI em alguns pacientes |
| Síndrome de ICF | Hipogamaglobulinemia, defeitos ocasionais leves das células T | Mutações no DNMT3B |
| Síndromes de Hiper-IgM | ||
| Ligadas ao X | Defeitos na célula B mediada pela célula T auxiliar, macrófago, e ativação das células dendríticas; defeitos na mutação somática, mudança de classe, e formação de centro germinativo; imunidade mediada por células defeituosas | Mutação no CD40L |
| Recessivo autossômico com defeitos imunológicos mediados por células | Defeitos na célula B mediada pela célula T auxiliar, macrófago, e ativação das células dendríticas; defeitos na mutação somática, mudança de classe, e formação de centro germinativo; imuni- dade mediada por células defeituosas | Mutações no CD40, NEMO |
| Recessivo autossômico com apenas defeito nos anticorpos | Defeitos na mutação somática e na mudança do isótipo | Mutações no AID, UNG |
AID, citidina deaminase induzida por ativação; DNMT3B, DNA metiltransferase 3B; ICF, anomalias faciais de instabilidade centromérica de imunodeficiências; ICOS, coestimulador induzível; NEMO, modulador essencial NF-κB; TACI, ativador da transmembrana e modulador de cálcio e interator ligante da ciclofilina; UNG, uracil N-glicosilase.
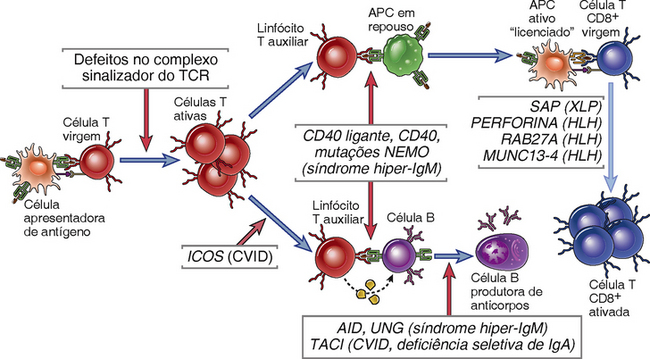
FIGURA 20-2 Imunodeficiência provocada pelos defeitos na ativação das células B e T. As imunodeficiências primárias podem ser provocadas por defeitos genéticos em moléculas necessárias para a sinalização do receptor de antígeno dos linfócitos T ou B, para a ativação de células B mediadas por células T auxiliares e APC, ou para a ativação de linfócitos T citotóxicos e células NK. CVID, imunodeficiência variável comum; HLH, linfo-histiocitose hemofagocítica.
Agamaglobulinemia Ligada ao X: Um Defeito de Sinalização do Pré-BCR Ligado ao X
A agamaglobulinemia ligada ao X, também chamada de agamaglobulinemiade é provocada por mutações ou deleções no gene que codifica uma enzima chamada Bruton tirosina cinase (Btk), que resulta em uma falha das células B de amadurecerem além do estágio da célula pré-B na medula óssea (Fig. 20-1). A doença é caracterizada pela ausência de gamaglobulina no sangue, como o nome indica. É uma das imunodeficiências congênitas mais comuns e o protótipo de uma falha da maturação das células B. A Btk está envolvida nos sinais de transdução do receptor das células pré-B (pré-BCR) que são necessários para a sobrevivência e diferenciação das células pré-B (Cap. 8). Em mulheres portadoras desta doença, apenas as células B que inativaram o cromossomo X carregando o alelo mutante amadurecem. Os pacientes com agamaglobulinemia ligada ao X geralmente têm Ig sérica baixa ou indetectável, células reduzidas ou ausentes no sangue periférico e tecidos linfoides, sem centros germinativos nos linfonodos, e sem células plasmáticas nos tecidos. A maturação, os números e as funções das células T são geralmente normais. Alguns estudos revelaram números reduzidos de células T ativadas nos pacientes, que pode ser uma consequência da apresentação dos antígenos reduzidos provocada pela falta de células B. Os distúrbios autoimunes se desenvolvem em quase 20% dos pacientes, por razões desconhecidas. As complicações infecciosas da agamaglobulinemia ligado ao X são muito reduzidas pelas injeções periódicas (semanal ou mensalmente) de preparações de gamaglobulina agrupadas. Estas preparações contêm anticorpos pré-formados contra os patógenos comuns e fornecem imunidade passiva eficaz.
Os camundongos Knockout que não tem Btk, bem como os camundongos Xid naturalmente mutantes por Btk, mostram um defeito menos grave na maturação das células B do que os seres humanos mostram, por que uma tirosina cinase semelhante à Btk chamada de Tec está ativa as células pré-B do rato que têm ausência de Btk e compensa parcialmente para a Btk mutante. As principais anormalidades nos camundongos Xid são as respostas dos anticorpos defeituosos a alguns antígenos polissacarídeos e uma deficiência no folicular maduro e nas células B-1 B.
Defeitos nos Pontos de Controle do Pré-BCR Recessivos Autossômicos
As formas recessivas autossômicas de agamaglobulinemia foram descritas, a maioria das quais pode estar ligada aos defeitos da sinalização pré-BCR. Os genes mutantes que foram identificados neste contexto incluem os genes que codificam a cadeia pesada μ (IgM), a cadeia leve substituta λ5, Igα (um componente de sinalização do pré-BCR e do BCR), e o BLNK (a jusante da proteína de um adaptador do pré-BCR e BCR).
Deficiências do Isótipo da Imunoglobulina Seletiva
Muitas imunodeficiências que envolvem seletivamente um ou alguns isótipos foram descritas. A mais comum é a deficiência de IgA seletiva, que afeta cerca de 1 de 700 caucasianos e é, portanto, a imunodeficiência primária mais comum conhecida. A deficiência de IgA geralmente ocorre esporadicamente, mas em muitos casos familiares ou com padrões de herança dominante autossômica ou recessiva autossômica também são conhecidos. As características clínicas são variáveis. Muitos pacientes são completamente normais; outros têm infecções respiratórias ocasionais e diarreia; e raramente, os pacientes têm infecções recorrentes graves que levam às lesões intestinais e das vias aéreas permanentes, com distúrbios autoimunes associados. A deficiência de IgA é caracterizada por baixos níveis séricos de IgA, geralmente menores que 50 μg/mL (normal, 2 a 4 mg/mL), com níveis normais ou elevados de IgM e IgG. O defeito nestes pacientes é um bloqueio na diferenciação das células B para as células plasmáticas secretoras de anticorpos IgA. Os genes de cadeia pesada α e a expressão do IgA associada às membranas são normais. Nenhuma anormalidade significante nos números, fenótipos ou respostas funcionais das células T foi observada nestes pacientes. Em uma pequena proporção de pacientes com deficiência de IgA seletiva, as mutações foram descritas no TACI (ativador da transmembrana, modulador de cálcio e interator do ligante da ciclofilina), um dos três tipos de receptores para as citocinas BAFF (fator de ativação das células B) e APRIL (um ligante indutor de proliferação). As mutações do TACI também são uma causa importante da imunodeficiência variável comum, discutidas posteriormente. A deficiência de IgA pode representar um fruste de forme da imunodeficiência variável comum.
As deficiências da subclasse IgG seletiva foram descritas em que os níveis séricos totais de IgG são normais, mas as concentrações de um ou mais subclasses estão abaixo do normal. A deficiência de IgG3 é a deficiência de subclasse mais comum nos adultos, e a deficiência de IgG2 associada à deficiência de IgA é a mais comum nas crianças. Alguns indivíduos com estas deficiências têm infecções bacterianas recorrentes, mas muitos não têm nenhum problema clínico. As deficiências da subclasse IgG seletiva se devem geralmente à diferenciação anormal das células B e raramente às exclusões homozigóticas de vários genes (Cγ) de regiões constantes.
Defeitos na Diferenciação das Células B: Imunodeficiência Variável Comum
A imunodeficiência variável comum é um grupo de distúrbios heterogêneos definido por níveis reduzidos de Ig sérica, respostas prejudicadas de anticorpos às infecções ou vacinas, e aumento da incidência de infecções. O diagnóstico geralmente é de exclusão quando outras doenças de imunodeficiência primária são descartadas. A apresentação e a patogênese são, como o nome indica, altamente variáveis. Embora a deficiência de Ig e as infecções piogênicas associadas tipicamente ao Haemophilus influenzae e o Streptococcus pneumoniae, sejam os principais componentes destes distúrbios, as doenças autoimunes, incluindo a anemia perniciosa, anemia hemolítica, doença intestinal inflamatória e artrite reumatoide, podem ser apenas significativas clinicamente. Uma alta incidência de tumores malignos, particularmente de linfomas, também está associada à imunodeficiência variável comum. Estes distúrbios podem ser diagnosticados precocemente na infância ou mais tarde durante a vida. Ambos os casos esporádicos e familiares ocorrem, o último com ambos os padrões de herança dominante e recessiva autossômica. Os linfócitos maduros B estão presentes nestes pacientes, mas as células plasmáticas estão ausentes nos tecidos linfoides, o que sugere um bloqueio na diferenciação de células B para as células secretoras de anticorpos. A produção de anticorpos defeituosos foi atribuída às anormalidades múltiplas, incluindo defeitos intrínsecos às células B, auxílio deficiente de células T e atividade excessiva da “célula supressora”. Uma pequena proporção de pacientes com imunodeficiência variável comum tem mutação no gene ICOS (coestimulador de células T induzíveis). O ICOS é necessário para a geração de células auxiliadoras foliculares T (Cap. 11). A causa mais comum desta síndrome é a existência de mutações no TACI, descritas anteriormente no contexto da deficiência do IgA seletivo. Alguns casos de imunodeficiência variável comum estão ligados às mutações no gene CD19. O CD19 é um componente de sinalização do complexo coreceptor do CR-2 (CD21) (Cap. 7).
Defeitos na Ativação das Células B Dependentes da Célula T: Síndromes de Hiper-IgM
A síndrome de hiper-IgM ligada ao X é provocada por mutações no gene que codifica a molécula efetora das células T do ligante CD40 (CD154). É um distúrbio raro associado à mudança defeituosa das células B para os isótipos IgG e IgA; estes anticorpos são, portanto, reduzidos, e o principal isotipo detectado no sangue é o IgM. As formas mutantes do ligante CD40 produzidas nestes pacientes não se ligam ou transduzem sinais através do CD40 e, portanto, não estimulam as células B a se submeter à mudança de isótipos de cadeia pesada, o que requer o auxílio de células T (Cap. 11). Os pacientes que sofrem de infecções semelhantes àquelas observadas em outras hipogamaglobulinemias. Os pacientes com síndrome de hiper-IgM ligado ao X também mostram defeitos na imunidade mediada por células, com aumento de suscetibilidade à infecção pelo micro-organismo fúngico intracelular Pneumocystis jiroveci. Esta imunidade defeituosa mediada por células ocorre por que o ligante CD40 também está envolvido na ativação dependente de células T dos macrófagos e das células dendríticas (Cap. 10). Os camundongos knockout que têm ausência de CD40 ou de ligante CD40 têm um fenótipo semelhante àquele da doença humana.
Casos raros da síndrome de hiper-IgM mostram um padrão de herança recessiva autossômica. Nestes pacientes, os defeitos genéticos podem estar no CD40 ou na deaminase induzida pela ativação das enzimas (AID), que está envolvida na mudança do isótipo da cadeia pesada e na mutação somática (Cap. 11). As mutações na AID são geralmente recessivas homozigóticas. Uma pequena fração das mutações na região do gene da AID, que corresponde à parte do C-terminal desta enzima apresenta um padrão de herança dominante autossômica. Uma forma de síndrome de hiper-IgM é provocada por mutações recessivas autossômicas no uracil N-glicosilase (UNG; Cap. 11), uma enzima que remove resíduos U dos genes Ig durante a mudança de classe e mutação somática. Um distúrbio hereditário, o EDA-ID, em que as mutações NEMO hipomórficas contribuem para um estado de hiper-IgM, assim como os defeitos nas estruturas ectodérmicas, são descritas anteriormente na seção sobre os defeitos de sinalização do TLR.
As mutações AID e UNG afetam a recombinação da mudança de classe e a hipermutação somática de maneiras distintas. Na ausência de AID, tanto a mudança como a hipermutação são defeituosas por que a AID é absolutamente necessária para ambos os processos. Na ausência de UNG, a mudança do isótipo é defeituosa, mas a hipermutação somática é grandemente preservada, embora apresente menos mutações A:T sem a atividade da UNG. O papel das mutações do gene de reparo do DNA nos defeitos de mudança de classe será considerado na seção sobre ataxia-telangiectasia posteriormente neste capítulo.
Defeitos na Ativação e Função dos Linfócitos T
As anormalidades congênitas na ativação dos linfócitos T estão sendo cada vez mais reconhecidas à medida que nossa compreensão da base molecular da ativação do linfócito melhora (Tabela 20-5). Incluídos nesta ampla categoria estão alguns distúrbios de composição granular das CTL e das células NK ou exocitose. Embora classificamos os distúrbios ligados à expressão MHC defeituosa com os distúrbios do desenvolvimento das células T, estas anormalidades também resultam na ativação defeituosa das células T que amadurecem e emergem do timo.
TABELA 20-5 Defeitos na Ativação da Célula T
| Doença | Deficiências Funcionais | Mecanismo de Defeito |
|---|---|---|
| Defeitos na expressão do MHC | ||
| Síndrome do linfócito essencial | Expressão e deficiência da MHC de classe II defeituosa nas células T CD4+; imunidade mediada por células defeituosas e respostas imunes humorais dependentes de T | Defeitos nos fatores de transcrição que regulam a expressão do gene da MHC de classe II, incluindo a CIITA, RFXANK, RFX5 e RFXAP |
| Deficiência de MHC de classe I | Redução dos níveis de MHC de classe I; células T CD8+ reduzidas | Mutações no TAP1, TAP2, e no TAPASIN |
| Sinalização defeituosa das células T | ||
| Defeitos de sinalização do TCR proximal | Defeitos na imunidade mediada por células e imunidade humoral dependente de T | Mutações nos genes CD3, CD45, STIM1, ORAI1 |
| Síndrome de Wiskott- Aldrich | Ativação das células T defeituosas, mobilidade dos leucócitos | Os rearranjos da actina-citoesqueleto dependentes de TCR são defeituosos devido às mutações no WASP |
| Linfo-histiocitose hemofagocítica familiar | ||
| Síndrome linfoproliferativa ligada ao X | Proliferação da célula B induzida pelo EBV descontrolado, macrófagos descontrolados e ativação da CTL, célula NK defeituosa e função CTL | Mutações no SAP |
| Deficiências da perforina | Mutações na PERFORINA | |
| Fusão dos grânulos | Exocitose defeituosa dos grânulos citotóxicos; mutações no RAB27A, MUNC13-4, SYNTAXIN, AP3 (e no LYST na síndrome de Chédiak-Higashi – Tabela 20-2) | |
AP3, complexo 3 da proteína relacionada ao adaptador; LYST, proteína reguladora do tráfico lisossômico; SAP, proteína associada ao SLAM; TAP, transportador associado ao processamento de antígenos; WASP, proteína da síndrome de Wiskott-Aldrich.
Defeitos na Transdução do Sinal TCR
Muitos exemplos de doenças de imunodeficiência rara provocadas por defeitos na expressão das moléculas necessárias para a ativação e função de células T foram identificados, e alguns já foram discutidos no contexto do SCID. As análises bioquímicas e moleculares dos indivíduos afetados revelaram mutações nos genes que codificam várias proteínas de células T (Tabela 20-5). Os exemplos incluem a expressão ou função do complexo TCR prejudicada, provocada por mutações nos genes CD3 ε ou γ, sinalização defeituosa mediada pelo TCR provocada por mutações no gene ZAP70, síntese reduzida das citocinas, tais como a IL-2 e a IFN-γ (em alguns casos provocada pelos defeitos nos fatores de transcrição), e ausência de expressão das cadeias do receptor IL-2. Estes defeitos são geralmente encontrados em apenas alguns casos ou em algumas famílias, e as características clínicas e a gravidade variam muito. Os pacientes com estas anomalias podem ter deficiências predominantemente na função das células T ou têm imunodeficiências misturadas nas células B e T, apesar dos números normais ou até mesmo dos números elevados dos linfócitos do sangue. Nós já consideramos previamente a importância do complexo CD3 no ponto de controle pré-TCR, o papel das mutações ZAP70 no desenvolvimento das células T CD8+, e a relevância do ORAI1 e do STIM1 e na ativação das células T, todos no contexto clínico do SCID. Outras síndromes que envolvem a ativação das células maduras T defeituosas são consideradas aqui.
Síndrome de Wiskott-Aldrich
Os graus variáveis de imunodeficiência das células T e B ocorrem em certas doenças congênitas com um amplo espectro de anormalidades que envolvem os sistemas de órgãos múltiplos. Um destes distúrbios é a síndrome de Wiskott-Aldrich, uma doença ligada ao X, caracterizada por eczema, trombocitopenia (plaquetas sanguíneas reduzidas) e suscetibilidade à infecção bacteriana. Algumas das anormalidades neste distúrbio podem ser atribuídas à ativação defeituosa das células T, embora a perda intrínseca da função das células B também contribua para a patogênese. Nos estágios iniciais da doença, os números de linfócitos são normais, e o defeito principal é a incapacidade de produzir anticorpos em resposta aos antígenos de polissacarídeos independentes de células T, por causa daqueles pacientes que são especialmente suscetíveis às infecções com bactérias encapsuladas. Os linfócitos (e plaquetas) são menores do que o normal. Com o aumento da idade, os pacientes apresentam número reduzido de linfócitos e imunodeficiência mais grave. O gene defeituoso responsável pela síndrome de Wiskott-Aldrich codifica uma proteína citoplasmática chamada WASP (proteína da síndrome de Wiskott-Aldrich), expressa exclusivamente nas células derivadas da medula óssea, que interagem com diversas proteínas, incluindo as moléculas adaptadoras do receptor do antígeno, tais como o Grb-2 (Cap. 7), o complexo Arp2/3 envolvido na polimerização de actina, e proteínas G pequenas da família Rho que regulam o rearranjo do citoesqueleto de actina. A ativação defeituosa e formação das sinapses nos linfócitos e a mobilidade defeituosa de todos os leucócitos podem ser responsáveis pela imunodeficiência observada nesta síndrome.
A Síndrome Linfoproliferativa Ligada ao X
A doença linfoproliferativa ligada ao X (XLP) é um distúrbio caracterizado pela incapacidade de eliminar o vírus Epstein-Barr (EBV), levando por fim à mononucleose infecciosa fulminante e ao desenvolvimento de tumores nas células B e à hipogamaglobulinemia associada. Em cerca de 80% dos casos, a doença se deve às mutações no gene que codifica a molécula de um adaptador chamada SAP (proteína associada ao SLAM) que se liga a uma família de moléculas da superfície celular envolvida na ativação das células NK e nos linfócitos T e B, incluindo a molécula de ativação do linfócito de sinalização (SLAM). O SAP se liga à SLAM das proteínas das membranas e ao 2B4 (Cap. 7) para a família Src do Fyn da cinase. Os defeitos no SAP contribuem para a ativação atenuada das células NK e T e resultam no aumento da suscetibilidade às infecções virais. Conforme discutido no Capítulo 11, o SAP é necessário para o desenvolvimento das células TFH, e a incapacidade dos pacientes XLP de gerar centros germinativos e anticorpos de alta afinidade provavelmente também contribui para a suscetibilidade à infecção viral. Em cerca de 20% dos casos do XLP, o defeito genético não reside no SAP, mas no gene que codifica o XIAP (inibidor ligado ao X da apoptose). A apoptose melhorada resultante das células T e as NKT leva a uma depleção acentuada destes tipos de células. Esta imunodeficiência é mais comumente manifestada por infecções EBV graves, que provavelmente surgem de forma oportunista, devido à natureza ubíqua do EBV.
Ativação das Células NK e da CTL Defeituosas: As Síndromes de Linfo-histiocitose Hemofagocíticas Familiares
As síndromes de linfo-histiocitose hemofagocítica (HLH) são um grupo de distúrbios de imunodeficiência fatal em que a secreção de grânulos da CTL e das células NK é defeituosa. Como resultado, as infecções virais não são mantidas sob controle, e a ativação do macrófago descontrolada é uma característica destas síndromes. Uma característica tardia, mas marcante destes distúrbios é a ingestão dos glóbulos vermelhos por macrófagos ativados (hemofagocitose). As mutações no gene da perforina, bem como as mutações nos genes que codificam a maquinaria celular envolvida na exocitose dos grânulos, podem contribuir para os fenótipos observados nesta síndrome. Especificamente, as mutações no RAB27A, uma pequena guanosina trifosfato envolvida na fusão vesicular, e no MUNC13-4, que codifica um adaptador que participa na exocitose dos grânulos, compromete a fusão dos grânulos líticos com a membrana plasmática e contribui, assim, com vários subtipos de HLH. Da mesma forma, as mutações no gene para um componente do complexo da proteína do adaptador citosólico AP-3 também podem interromper o transporte intracelular e contribuir para uma forma de HLH. Acredita-se que as células T e macrófagos respondem fortemente aos micro-organismos para compensar pelos defeitos das células CTL e NK, e estas respostas compensatórias são manifestas pela hemofagocitose e pela linfadenopatia no contexto da imunodeficiência.
Distúrbios Multissistêmicos com Imunodeficiência: Ataxia-Telangiectasia
A imunodeficiência é muitas vezes parte de uma constelação de sintomas em uma série de doenças hereditárias. Exemplos destas síndromes discutidas anteriormente incluem a síndrome de Chédiak-Higashi, síndrome de Wiskott-Aldrich, e síndrome de DiGeorge. A ataxia-telangiectasia é um distúrbio recessivo autossômico caracterizado por movimentação anormal (ataxia), malformações vasculares (telangiectases), déficits neurológicos, aumento da incidência de tumores e imunodeficiência. Os defeitos imunológicos são de gravidade variável e podem afetar tanto as células B como as células T. Os defeitos imunológicos humorais mais comuns são a deficiência de IgA e IgG2, provavelmente devido ao papel crucial que a proteína da ATM desempenha na recombinação da mudança de classe (discutido posteriormente). Os defeitos das células T, que são geralmente menos pronunciados, estão associados à hipoplasia do tipo. Os pacientes experimentam infecções bacterianas no trato respiratório inferior e superior, múltiplos fenômenos autoimunes e câncer cada vez mais frequente com o avanço da idade. O gene responsável por este distúrbio está localizado no cromossomo 11 e codifica uma proteína chamada ATM (ataxia-telangiectasia modificada) que está relacionada, estruturalmente, ao fosfatidilinositol 3-cinase, mas é uma proteína cinase. A proteína ATM pode ativar pontos de controle do ciclo celular e a apoptose em resposta às quebras de fitas duplas de DNA e também mostrou contribuir para a estabilidade dos complexos de quebra das fitas duplas de DNA durante a recombinação de V(D)J. Devido a estas anormalidades no reparo do DNA, a geração dos receptores de antígenos também pode ser anormal.
O reparo do DNA durante a recombinação da mudança de classe não apenas envolve a via de junção final não homóloga, mas também requer a proteína ATM, a proteína MRE11 (a recombinação meiótica 11), e a proteína NBS1 (síndrome do ponto de parda Nijmegen 1). Os pacientes com mutações nos genes que codificam estas proteínas ou ATM muitas vezes apresentam redução dos níveis de IgG, IgA e IgE.
Abordagens Terapêuticas para as Imunodeficiências Congênitas
O tratamento atual das imunodeficiências tem dois objetivos: minimizar e controlar as infecções e substituir os componentes defeituosos ou ausentes do sistema imune através da transferência adotiva ou transplante. A imunização passiva com gamaglobulina agrupada é muito benéfica para pacientes agamaglobulinêmicos e salvou a vida de muitos meninos com agamaglobulinemia ligada ao X. O transplante de células-tronco hematopoiéticas é atualmente o tratamento de escolha para muitas doenças de imunodeficiência e tem sido bem-sucedido no tratamento do SCID com deficiência de ADA, síndrome de Wiskott-Aldrich, síndrome de linfócitos essenciais e deficiências de adesão de leucócitos. É mais bem-sucedido com depleção cuidadosa de células T da medula óssea e combinação de HLA para evitar a doença do hospedeiro versus enxerto (Cap. 16). A terapia de substituição de enzimas para a ADA e deficiências de PNP tem sido tentada, com transfusões de glóbulos vermelhos utilizados como fonte das enzimas. Esta abordagem produziu melhora clínica temporária em vários pacientes com SCID autossômico. A injeção de ADA em bovinos conjugada a polietilenoglicol para prolongar sua meia-vida sérica tem sido bem-sucedida em alguns casos, mas os benefícios geralmente são de curta duração.
Em teoria, a terapia de escolha para distúrbios congênitos dos linfócitos é substituir o gene defeituoso nas células-tronco autorrenováveis. A substituição do gene continua uma meta distante para a maioria das imunodeficiências no presente, apesar dos esforços consideráveis. Os principais obstáculos para este tipo de terapia genética são as dificuldades para purificar as células-tronco autorrenováveis, que são o alvo ideal para a introdução do gene de substituição, e na introdução nas células para alcançar expressão estável, de longa duração e de alto nível. Tem sido feito algum progresso na terapia genética para a deficiência de ADA pelo uso de uma abordagem de condicionamento mais suave para esgotar as células hospedeiras da medula óssea, o que facilita o enxerto e a proliferação de células-tronco modificadas introduzidas no hospedeiro. Um pequeno número de pacientes com SCID ligado ao X foi tratado de forma bem-sucedida através do transplante de células da medula óssea autóloga projetada para expressar um gene γc normal. No entanto, alguns pacientes tratados desenvolveram leucemia, aparentemente por que o gene γc introduzido inseriu adjacências a um oncogêne e ativou este gene. Como resultado, o futuro da terapia genética para esta doença é incerto.
IMUNODEFICIÊNCIAS ADQUIRIDAS (SECUNDÁRIAS)
As deficiências do sistema imune muitas vezes se desenvolvem devido às anormalidades que não são genéticas, mas adquiridas durante a vida (Tabela 20-6). A mais proeminente destas anormalidades é a infecção pelo HIV, e isto é descrito na próxima seção. As doenças de imunodeficiência adquirida são provocadas por dois tipos de mecanismos patogênicos. Primeiro, a imunossupressão pode ocorrer como uma complicação biológica de outro processo de doença. Segundo, as chamadas imunodeficiências iatrogênicas podem se desenvolver como complicações da terapia de outras doenças.
TABELA 20-6 Imunodeficiências Adquiridas
| Causa | Mecanismo |
|---|---|
| Infecção por HIV | Depleção das células T CD4+ |
| Desnutrição proteico-calórica | Os transtornos metabólicos inibem a maturação e função dos linfócitos |
| Radiação e quimioterapia para o câncer | Diminuição dos precursores dos linfócitos da medula óssea |
| Metástases de câncer e leucemia envolvendo a medula óssea | Local reduzido de desenvolvimento de leucócitos |
| Imunossupressão para transplantes, doenças autoimunes | Ativação de linfócitos reduzida |
| Remoção do baço | Fagocitose de micro- organismos diminuída |
As doenças nas quais a imunodeficiência é um elemento de complicação comum incluem a desnutrição, as neoplasias e as infecções. A desnutrição proteico-calórica é comum nos países em desenvolvimento e está associada à imunidade humoral e celular dos micro-organismos prejudicada. Grande parte da morbidade e mortalidade que afligem as pessoas desnutridas se deve às infecções. A base para a imunodeficiência não está bem definida, mas é razoável supor que os distúrbios metabólicos globais nestes indivíduos, provocados pela ingestão insuficiente de proteína, gordura, vitaminas e minerais, afetará negativamente a maturação e função das células do sistema imunológico.
Os pacientes com câncer bem avançado são geralmente suscetíveis à infecção devido às respostas imunes humorais e mediadas por células a uma variedade de organismos prejudicadas. Os tumores da medula óssea, incluindo cânceres metásticos de medula e leucemias que surgem na medula, podem interferir no crescimento e desenvolvimento de linfócitos normais e outros leucócitos. Além disso, os tumores podem produzir substâncias que interferem no desenvolvimento ou função dos linfócitos. Um exemplo de imunodeficiência associada à malignidade é a deficiência na função das células T comumente observadas nos pacientes com um tipo de linfoma chamado doença de Hodgkin. Este defeito foi primeiro caracterizado como uma incapacidade de produzir uma reação DTH na injeção intradérmica de vários antígenos comuns aos quais os pacientes foram expostos previamente, tais como Candida ou toxoide tetânico. Outras medidas in vitro da função das células T, tais como respostas proliferativas a ativadores policlonais, também são prejudicadas nos pacientes com doença de Hodgkin. Esta doença generalizada nas respostas DTH é chamada de anergia. A causa das anormalidades destas células T é desconhecida.
Vários tipos de infecções levam à imunossupressão. Outros vírus diferentes do HIV são conhecidos por prejudicar as respostas imunológicas; exemplos incluem o vírus do sarampo e o vírus linfotrópico das células T humanas (HTLV-1). Ambos os vírus podem infectar os linfócitos, que pode ser uma base para seus efeitos imunossupressores. Como o HIV, o HTLV-1 é um retrovírus com tropismo para as células T CD4+; no entanto, ao invés de matar as células T auxiliares, ele as transforma e produz um neoplasma maligno agressivo das células T chamado leucemia/linfoma das células T adultas (ATL). Os pacientes com ATL normalmente têm imunossupressão grave, com infecções oportunistas múltiplas. As infecções crônicas com Mycobacterium tuberculosis e vários fungos geralmente resultam em anergia para muitos antígenos. As infecções parasitárias crônicas também podem levar à imunossupressão. Por exemplo, as crianças africanas com infecções de malária crônica têm a função das células T deprimida, esta pode ser uma das razões pela qual estas crianças têm uma propensão maior a desenvolver tumores malignos associados à EBV.
As imunossupressões iatrogênicas são mais frequentes devido às terapias medicamentosas que matam ou inativam funcionalmente os linfócitos. Alguns medicamentos são ministrados intencionalmente como imunossupressores, tanto pra o tratamento de doenças inflamatórias ou para evitar a rejeição do transplante de órgãos. Os medicamentos anti-inflamatórios e imunossupressores mais comumente usados são os corticosteroides e a ciclosporina, respectivamente. Vários medicamentos quimioterápicos são administrados a pacientes com câncer, e estes medicamentos são geralmente citotóxicos para os linfócitos maduros e em desenvolvimento, bem como para os precursores dos granulócitos e monócitos. Portanto, a quimioterapia do câncer é quase sempre acompanhada por um período de imunossupressão e risco de infecção. A imunossupressão iatrogênica e os tumores que envolvem a medula óssea são as causas mais comuns de imunodeficiência nos países desenvolvidos.
Outra forma de imunossupressão adquirida resulta da ausência do baço provocada por remoção cirúrgica do órgão após trauma e como tratamento de certas doenças hematológicas ou por infarto na doença falciforme. Os pacientes sem baço são mais suscetíveis à infecção por alguns organismos, principalmente por bactérias encapsuladas, como o Streptococcus pneumoniae. Esta suscetibilidade maior se deve em parte à remoção fagocitária do defeito dos micro-organismos transmitidos pelo sangue opsonizado, uma importante função fisiológica do baço, e em parte devido às respostas defeituosas dos anticorpos resultantes da ausência de células B na zona marginal.
VÍRUS DA IMUNODEFICIÊNCIA HUMANA E SÍNDROME DA IMUNODEFICIÊNCIA ADQUIRIDA
A AIDS é a doença provocada pela infecção por HIV e é caracterizada pela imunossupressão profunda com infecções oportunistas associadas e tumores malignos, desperdício e degeneração do sistema do nervoso central (SNC). O HIV infecta uma variedade de células do sistema imunológico, incluindo as células T CD4+ auxiliares, macrófagos e células dendríticas. O HIV evoluiu como um patógeno humano muito recentemente em relação à maioria dos outros patógenos humanos conhecidos, e o HIV epidêmico foi identificado pela primeira vez apenas nos anos oitenta. No entanto, o grau de morbidade e mortalidade provocado pelo HIV e o impacto global da infecção por HIV nos recursos de saúde e a economia já são imensos e continuam a crescer. O HIV já infectou 50 a 60 milhões de pessoas e provocou a morte de mais de 22 milhões de adultos e crianças. Cerca de 35 milhões de pessoas vivem com infecção por HIV e AIDS, dos quais aproximadamente 70% estão na África e 20% na Ásia, e quase 2 milhões morrem da doença todos os anos. A doença é especialmente devastadora porque cerca de metade dos cerca de 3 milhões de novos casos a cada ano ocorrem em jovens adultos (15-24 anos). A AIDS deixou aproximadamente 14 milhões de órfãos e resultou na morte de aproximadamente 30 milhões de pessoas. Atualmente não existe vacina eficaz ou cura para a AIDS, mas terapias antirretrovirais bastante eficazes têm sido desenvolvidas. Nesta seção do capítulo, descrevemos as propriedades moleculares e biológicas do HIV, a patogênese da imunodeficiência induzida pelo HIV, e a características clínicas e epidemiológicas de doenças relacionadas ao HIV.
Características Moleculares e Biológicas do HIV
O HIV é um membro da família lentivírus do retrovírus animal. O lentivírus, incluindo o vírus visna dos ovinos e os vírus de imunodeficiência dos bovinos, felinos e símios, é capaz de infecções nas células latentes a longo prazo e efeitos citopáticos em curto prazo, e todos eles produzem doenças fatais que progridem lentamente, que incluem síndromes de desperdício e degeneração do SNC. Dois tipos de HIV estreitamente relacionados, designados como HIV-1 e HIV-2, foram identificados. O HIV-1 é de longe a causa mais comum da AIDS; o HIV-2, que difere em estrutura genômica e antigenicidade, provoca uma forma de AIDS com progressão mais lenta do que as doenças ligadas ao HIV-1.
Estruturas e Genes do HIV
Uma partícula infecciosa do HIV consiste em duas fitas idênticas de RNA embaladas com um núcleo de proteínas virais e envoltas por um envelope de duas camadas de fosfolipídios derivado da membrana da célula hospedeira, mas incluindo as proteínas da membrana viralmente codificada (Fig. 20-3). O genoma RNA do HIV tem aproximadamente 9,2 kb de comprimento e tem o arranjo básico da característica das sequências de ácido nucleico de todos os retrovírus conhecidos (Fig. 20-4). As repetições de terminais longos (LTRs) em cada extremidade do genoma regulam e expressão genética viral, a integração viral no genoma hospedeiro e a replicação viral. A sequência gag codifica as proteínas estruturais do núcleo. A sequência env codifica as glicoproteínas gp120 e gp41 do envelope, que se associam de forma não covalente umas às outras e são necessárias para a infecção das células. A sequência pol codifica a transcriptase reversa, a integrase e as enzimas da protease viral necessárias para a replicação viral. Além destes típicos genes retrovírus, o HIV-1 também inclui seis outros genes reguladores, nomeadamente, os genes tat, rev, vif, nef, vpr e o vpu, cujos produtos regulam a reprodução viral e a evasão imunológica do hospedeiro de várias maneiras. As funções destes genes estão resumidas na Figura 20-4.
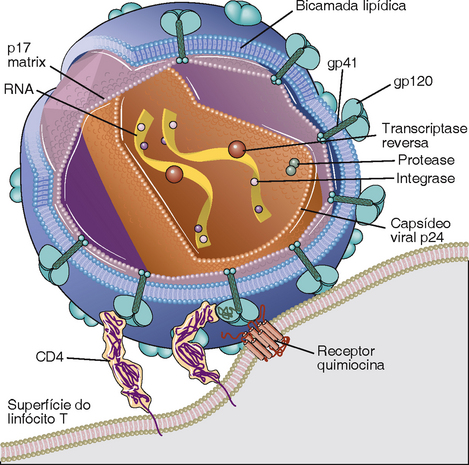
FIGURA 20-3 Estrutura do HIV-1. Um vírion de HIV-1 é mostrado próximo de uma superfície de células T. O HIV-1 consiste em duas fitas idênticas de RNA (o genoma viral) e enzimas associadas, incluindo a transcriptase reversa, integrase e protease, envoltas em um núcleo em forma de cone composto de proteína capsídeo p24, rodeada por uma matriz de proteína p17, tudo envolto por um envelope de membrana fosfolipídica derivado de uma célula hospedeira. As proteínas da membrana codificada viralmente (gp41 e gp120) são vinculadas ao envelope. O CD4 e os receptores de quimiocinas na função de superfície da célula hospedeira como receptores do HIV-1.
(© 2000 Terese Winslow.)
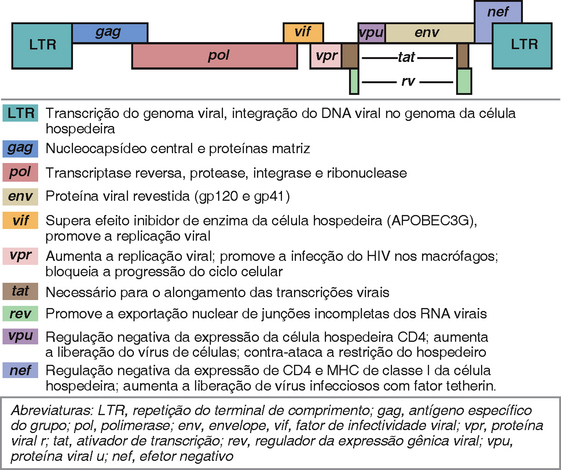
FIGURA 20-4 Genoma do HIV-1. Os genes junto com o genoma linear são indicados como blocos coloridos de forma diferente. Alguns genes usam algumas das mesmas sequências de outros genes, conforme mostrado pelos blocos sobrepostos, mas são lidos de forma diferente pela polimerase do RNA das células hospedeiras. Da mesma forma, os blocos separados por linhas indicam genes cujas sequências codificadas são separadas no genoma e necessitam da união do RNA para produzir mRNA funcional.
(Modificado de Greene W. AIDS and the immune system. Copyright 1993 by Scientific American, Inc. Todos os direitos reservados.)
Ciclo de Vida Viral
A infecção por HIV das células começa quando a glicoproteína do envelope (Env) do vírus se liga tanto ao CD4 como a um correceptor que é um membro da família dos receptores de quimiocinas (Fig. 20-5). As partículas virais que iniciam a infecção estão geralmente no sangue, sêmen, ou outros fluidos corporais de um indivíduo e são introduzidas em outro indivíduo por contato sexual, através de agulha, ou passagem transplacentária. O Env é um complexo composto por uma subunidade transmembranar gp41 e uma subunidade externa, associada de forma não covalente gp120. Estas subunidades são produzidas por clivagem proteolítica de um precursor gp160. O complexo Env é expresso como uma estrutura trimérica de três pares de gp120/gp41. Este complexo medeia um processo de várias etapas de fusão do envelope virion com a membrana da célula-alvo (Fig. 20-6). O primeiro passo deste processo é a ligação das subunidades gp120 às moléculas CD4, que induz a uma mudança conformacional que promove a ligação secundária do gp120 a um coreceptor de quimiocina. A ligação do coreceptor induz a uma mudança conformacional na gp41 que expõe uma região hidrofóbica, chamada de peptídeo de fusão, que se insere na membrana celular e permite que a membrana viral se funda com a membrana da célula-alvo. Após o vírus completar seu ciclo de vida na célula infectada (descrito posteriormente), as partículas virais livres são liberadas de uma célula infectada e se ligam a uma célula não infectada, propagando, assim, a infecção. Além disso, a gp120 e a gp41, que são expressas na membrana plasmática de células infectadas antes que o vírus seja liberado, podem mediar à fusão da célula-célula com uma célula não infectada que expressa o CD4 e os coreceptores, e os genomas podem então ser transmitidos entre as células fundidas diretamente.
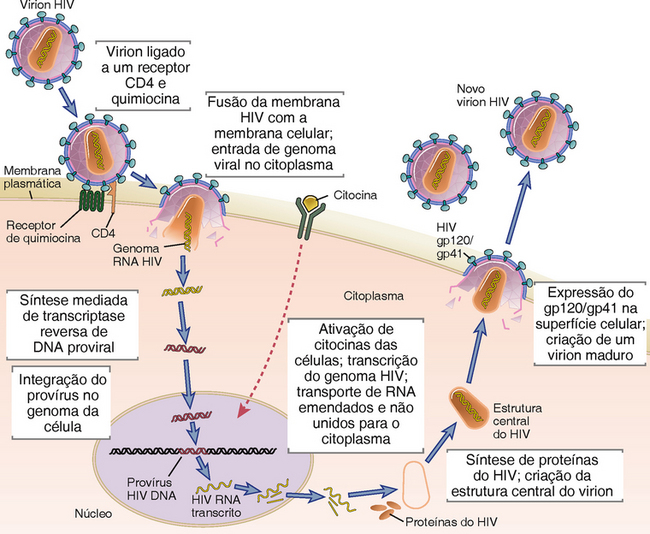
FIGURA 20-5 Ciclo de vida do HIV. Os passos sequênciais no ciclo de vida do HIV são mostrados, a infecção inicial de uma célula hospedeira para replicação viral e lançamento de um novo virion. Por uma questão de clareza, a produção e liberação de apenas um novo virion são mostrados. Uma célula infectada produz realmente muitos virions, cada um capaz de infectar as células, aumentando assim o ciclo infeccioso.
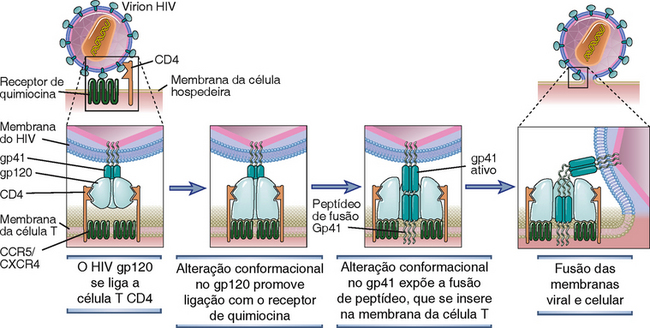
FIGURA 20-6 Mecanismos de entrada do HIV em uma célula. No modelo descrito, as mudanças conformacionais sequênciais no gp120 e no gp 41 são induzidas pela ligação a um CD4. Estas mudanças promovem a ligação do vírus ao correceptor (um receptor de quimiocinas) e a fusão do HIV-1 e das membranas celulares hospedeiras. O peptídeo de fusão do gp41 ativado contém resíduos de aminoácidos que mediam a inserção na membrana plasmática das células hospedeiras.
Os receptores de quimiocinas mais importantes que agem como correceptores para HIV são o CXCR4 e o CCR5. Mais de sete receptores de quimiocinas diferentes foram mostrados para servir como coreceptores para a entrada do HIV nas células, e diversas outras proteínas que pertencem à proteína G que abrange as sete transmembranas, acopladas à família de receptores, como o receptor do leucotrieno B4, também podem mediar à infecção por HIV das células. Isolados diferentes de HIV têm tropismos distintos para diferentes populações de células que estão relacionadas com a especificidade de variantes de gp120 para diferentes receptores de quimiocinas. Todas as cepas do HIV podem infectar e replicar nas células T CD4+ humanas recém-isoladas que são ativadas in vitro. Em contraste, algumas células infectarão culturas primárias de macrófagos humanos, mas não linhas contínuas de células T (macrófagos-tróficos, ou vírus M-tróficos), enquanto outras cepas infectarão linhas de células T, mas não os macrófagos (vírus T-tróficos). Algumas cepas de vírus também infectam tanto as linhas das células T como os macrófagos (vírus dual-trófico). O isolamento do vírus trófico macrófago expressa uma gp120 que se liga ao CCR5, que é expresso nos macrófagos (e algumas células T de memória), enquanto os vírus tróficos das células T se ligam ao CXCR4, que é expresso nas linhas das células T. Os variantes do HIV são descritos como X4 para a ligação CXCR, R5 para a ligação CCR5, ou R5X4 para a capacidade de se ligar a ambos os receptores de quimiocinas. Em muitos indivíduos infectados pelo HIV, há uma mudança da produção do vírus que usa o CCR5 e é predominantemente trófico-macrófago no início da doença para o vírus que se liga ao CXCR4 e é trófico-linha para a célula T no final da doença. As cepas dos T- tróficos tendem a ser mais virulentas, presumivelmente porque elas infectam e destroem as células T mais do que as cepas M-trófico. A importância do CCR5 na infecção por HIV in vivo é apoiada pela descoberta de que os indivíduos que não expressam este receptor na superfície celular devido à eliminação de um homozigoto herdado 32-bp no gene CCR5 são resistentes à infecção por HIV.
Uma vez que um virion HIV entra em uma célula, as enzimas dentro do complexo da nucleoproteína se tornam ativas e começam o ciclo reprodutivo viral (Fig. 20-5). O núcleo da nucleoproteína do vírus se torna interrompido, o genoma RNA do HIV é transcrito reversamente para uma forma de DNA de fita dupla pela transcriptase reversa viral, e o DNA viral entra no núcleo. A integrase viral também entra no núcleo e catalisa a integração do DNA viral no genoma da célula hospedeira. O DNA do HIV integrado é chamado de provírus. O provírus pode permanecer inativo de forma transcricional por meses ou anos, com pouca ou nenhuma produção de novas proteínas virais ou virions, e desta forma a infecção por HIV de uma célula individual pode ser latente.
A transcrição dos gene do pró-vírus do DNA integrado é regulamentada pelo montante de genes estruturais virais do LTR, e as citocinas e outros estímulos fisiológicos que ativam as células T e os macrófagos melhoram a transcrição dos genes virais. Os LTR contêm sequências de sinais de poliadenilação, a sequência promotora de caixas TATA, e os locais de ligação para dois fatores de transcrição de células hospedeiras, NF-κB e SP1. O início da transcrição do gene do HIV nas células T está ligado à ativação das células T pelo antígeno ou pelas citocinas. Por exemplo, os ativadores policlonais das células T, como a fito hemaglutinina, e as citocinas como a IL-2, o fator de necrose tumoral (TNF), e a linfotoxina estimulam a expressão genética do HIV nas células T infectadas, e o IL-1, IL-3, IL-6, TNF, a linfotoxina, IFN-γ e o fator estimulante da colônia granulócitos-macrófagos (GM-CSF) estimulam a expressão genética do HIV e a replicação viral nos monócitos e macrófagos infectados. O TCR e o estímulo de citocina da transcrição genética do HIV provavelmente envolvem a ativação do NF-κB e sua ligação às sequências no LTR. Este fenômeno é significativo para a patogênese da AIDS por que a resposta normal de uma célula T infectada de forma latente a um micro-organismo pode ser o caminho em que a latência é encerrada e a produção de vírus comece. As infecções múltiplas que os pacientes com AIDS adquirem, portanto, estimulam a produção de HIV e a infecção das células adicionais.
A proteína Tat é necessária para a expressão genética do HIV e age aumentando a produção das transcrições virais completas de mRNA. Mesmo na presença de sinais ideais para iniciar a transcrição, poucas moléculas do mRNA do HIV são realmente sintetizadas sem a ação do TAT por que a transcrição dos genes do HIV pela polimerase do RNA dos mamíferos é ineficiente e o complexo da polimerase geralmente pára antes do mRNA ser concluído. A proteína Tat se liga ao mRNA nascente e aumenta a “processabilidade” da polimerase do RNA em centenas de vezes, o que permite que a conclusão da transcrição produza um mRNA viral funcional.
A síntese das partículas virais infecciosas maduras começa depois que as transcrições completas do RNA viral são produzidas e os genes virais são expressos como proteínas. Os mRNA que codificam as diversas proteínas do HIV são provenientes de uma transcrição integral do genoma por eventos de ligação diferencial. A expressão genética do HIV pode ser dividida em um estágio inicial, durante o qual os genes reguladores são expressos, e uma fase final, durante a qual os genes estruturais são expressos e os genomas virais completos são embalados. As proteínas Rev, Tat e Nef são produtos genéticos iniciais codificados por mRNAs ligados completamente que são exportados do núcleo e traduzidos em proteínas no citoplasma logo após a infecção de uma célula. Os genes tardios incluem o env, gag e pol, que codificam os componentes estruturais dos vírus e são traduzidos de RNA ligados isoladamente ou separados. A proteína Rev inicia a mudança do início para o fim da expressão genética promovendo a exportação destes últimos RNA genéticos ligados incompletamente para fora do núcleo. O produto genético pol é uma proteína precursora que é clivada sequencialmente para formar transcriptase reversa, protease, ribonuclease e enzimas de integrase. Conforme mencionado anteriormente, a transcriptase reversa e as proteínas da integrase são necessárias para produzir uma cópia do DNA do genoma do RNA viral e para integrá-lo como um pró-vírus no genoma do hospedeiro. O gene gag codifica uma proteína 55 kD que é proteoliticamente clivada nos polipeptídeos p24, p17 e p15 através da ação da protease viral codificada pelo gene pol. Estes polipeptídeos são as proteínas nucleares que são necessárias para a montagem de partículas virais infecciosas. O produto primário do gene env é uma glicoproteína 160 kD (gp160) que é clivada por proteases celulares dentro do retículo endoplasmático nas proteínas gp120 e gp41 necessárias para a ligação do HIV com as células, conforme discutido anteriormente. A terapia medicamentosa antiviral atual para doenças com HIV inclui inibidores das enzimas transcriptase reversa, protease e integrase.
Após a transcrição de vários genes virais, as proteínas virais são sintetizadas no citoplasma. A montagem de partículas virais infecciosas então começa através de transcrições de RNA completos do genoma proviral dentro de um complexo nucleoproteico que inclui as proteínas nucleares do gag e as enzimas codificadas do pol necessárias para o próximo ciclo de integração. Este complexo nucleoprotéico, então, prolifera da membrana plasmática, capturando o Env e as glicoproteínas do hospedeiro como parte de seu envelope. A taxa de produção do vírus pode chegar a níveis suficientemente altos para provocar a morte celular, conforme discutido posteriormente.
Um fator do hospedeiro que impede o virion de libertar certos tipos de células é uma proteína chamada tetherin. A tetherin impede o pinçamento de determinados vírus, incluindo o HIV, e sua inibição do processo de integração pode ser antagonizado por uma proteína do HIV chamada Vpu.
Patogênese da Infecção pelo HIV e pela AIDS
A doença do HIV começa com uma infecção aguda, que é apenas parcialmente controlada pela resposta imune adaptativa, e avança para a infecção progressiva crônica dos tecidos linfoides periféricos (Fig. 20-7). O vírus geralmente entra através do epitélio da mucosa. Os eventos subsequentes na infecção podem ser divididos em várias fases.
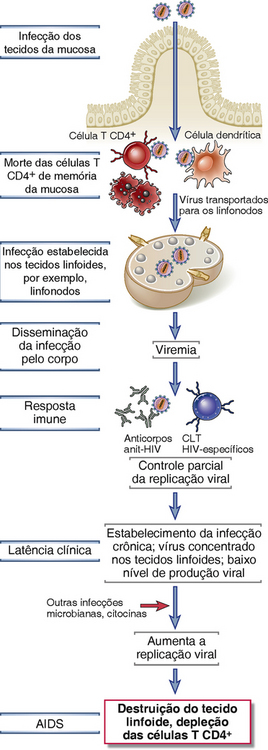
FIGURA 20-7 Progressão da infecção pelo HIV. A progressão da infecção pelo HIV está relacionada com a disseminação do vírus do local inicial da infecção para os tecidos linfoides através do corpo. A resposta imune do hospedeiro controla temporariamente a infecção aguda, mas não impede o estabelecimento da infecção crônica das células nos tecidos linfoides. Os estímulos das citocinas induzidos por outros micro-organismos servem para melhorar a produção de HIV e a progressão para a AIDS.
A infecção aguda (inicial) é caracterizada pela infecção das células da memória T CD4+ nos tecidos linfoides das mucosas e na morte de muitas células infectadas. Como os tecidos da mucosa são o maior reservatório de células T no corpo e o principal local de residência das células T da memória, esta perda local é refletida na diminuição considerável dos linfócitos. Na verdade, dentro de 2 semanas de infecção, uma grande facção de células T CD4+ pode ser destruída.
A transição da fase aguda para uma fase crônica da infecção é caracterizada pela disseminação do vírus, viremia e pelo desenvolvimento de respostas imunes do hospedeiro. As células dendríticas no epitélio nos locais de entrada de vírus capturam o vírus e então migram para os nódulos linfáticos. As células dendríticas expressam uma proteína com um domínio de lectina que se liga à manose, chamado DC-SIGN, que pode ser particularmente importante na ligação do envelope HIV e no transporte de vírus. Uma vez nos tecidos linfoides, as células dendríticas podem transmitir o HIV para as células T CD4+ através do contato direto célula-célula. Dentro de poucos dias após a primeira exposição ao HIV, a replicação viral pode ser detectada nos linfonodos. Esta replicação leva à viremia, durante a qual um grande número de partículas de HIV está presentes no sangue do paciente, acompanhadas por uma síndrome aguda de HIV, que inclui uma variedade de sinais não específicos e sintomas típicos de muitas infecções virais (descritos posteriormente). A viremia permite que o sangue se dissemine por todo o corpo e infecte as células T auxiliares, os macrófagos e as células dendríticas nos tecidos linfoides periféricos. À medida que a infecção do HIV se espalha, o sistema imunológico adaptativo estabelece tanto as respostas imunológicas mediadas por células como as humorais direcionadas aos antígenos virias, que serão descritas posteriormente. Estas respostas imunológicas controlam parcialmente a infecção e a produção viral, e este controle é refletido por uma queda na viremia para níveis baixos, mas detectáveis de aproximadamente 12 semanas após a exposição primária.
Na próxima fase crônica da doença, os linfonodos e o baço são locais de replicação de contínua de HIV e destruição celular (Fig. 20-7). Durante este período da doença, o sistema imunológico permanece competente em lidar com a maioria das infecções com micro-organismos oportunistas, e poucas ou nenhuma manifestação clínica da infecção por HIV estão presentes. Portanto, esta fase da doença por HIV é chamada de período de latência clínica. Embora a maioria de células T no sangue periférico não abrigue o vírus, a destruição das células T CD4+ dentro dos tecidos linfoides progride de forma constante durante o período latente, e o número de células T CD4+ do sangue circulante diminui de forma constante (Fig. 20-8). Mais de 90% de aproximadamente 1012 células T do corpo são encontradas normalmente nos tecidos linfoides periféricos e da mucosa, e estima-se que o HIV destrua até 1 a 2 × 109 células T CD4+ todos os dias. No início do curso da doença, o organismo pode continuar a produzir novas células T CD4+ e, portanto, essas células podem ser substituídas quase tão rapidamente quanto elas são destruídas. Nesta fase, até 10% das células T CD4+ nos órgãos linfoides podem ser infectadas, mas o número de células T CD4+ em circulação que são infectadas ao mesmo tempo pode ser inferior a 0,1% do total de células T CD4+ em um indivíduo. Por fim, durante alguns anos, o ciclo contínuo de infecção por vírus, a morte das células T, e as novas infecções levam a uma diminuição constante no número de células T CD4+ nos tecidos linfoides e na circulação.
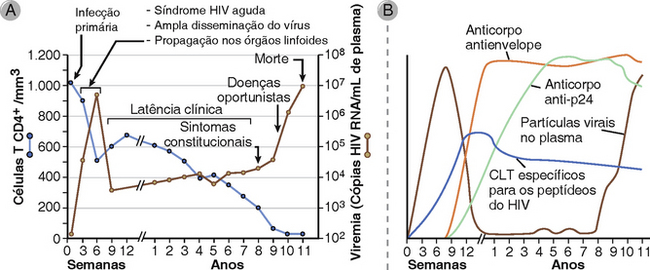
FIGURA 20-8 Curso clínico do HIV. A, Plasma viremia, contagem de células T CD4+ sanguíneas e estágios clínicos da doença. Cerca de 12 semanas após a infecção, o vírus transmitido pelo sangue (plasma viremia) é reduzido a níveis muito baixos (detectáveis apenas por ensaios de reações em cadeia de polimerase-transcriptase reversa sensível) e permanece assim por muitos anos. No entanto, a contagem de células T CD4+ sofre redução constante durante este período de latência clínica devido à replicação viral ativa e à infecção das células T nos linfonodos. Quando a contagem das células T CD4+ cai abaixo de um nível crítico (cerca de 200/mm3), o risco de infecção e outras características clínicas de AIDS é alto. B, Resposta imune à infecção por HIV. Uma resposta CTL ao HIV é detectável de 2 a 3 semanas após a infecção inicial e picos de 9 a 12 semanas. A forte expansão das células T CD8+ de vírus específicos ocorre durante este tempo, e até 10% das CTL de um paciente podem ser específicas de HIV em 12 semanas. A resposta imune humoral atinge seu pico de HIV em cerca de 12 semanas.
(De Pantaleo G, C Graziosi, and AS Fauci. New concepts in the immunopathogenesis of human immunodeficiency virus infection. New England Journal of Medicine 328:327-335, 1993. Copyright © 1993 Massachusetts Medical Society. Todos os direitos reservados.)
Mecanismos de Imunodeficiência Provocados pelo HIV
A infecção por HIV, em última análise resulta na função prejudicada de ambos os sistemas imunológicos adaptativo e inato. Os defeitos mais proeminentes estão na imunidade mediada pelas células, e eles podem ser atribuídos a diversos mecanismos, incluindo os efeitos citopáticos diretos do vírus e dos efeitos indiretos.
Uma causa importante da perda de células T CD4+ nas pessoas infectadas pelo HIV é o efeito citopático direto da infecção destas células pelo HIV. A morte das células T CD4+ está associada à produção do vírus nas células infectadas e é uma das principais causas da redução nos números destas células, especialmente na fase inicial (aguda) da infecção. Diversos efeitos tóxicos diretos do HIV nas células CD4+ infectadas foram descritos.
• O processo de produção de vírus, com a expressão do gp41 na membrana plasmática e do surgimento de partículas virais, pode levar ao aumento da permeabilidade da membrana plasmática e ao influxo das quantidades letais de cálcio, o que induz a apoptose, ou à lise osmótica da célula provocada pelo influxo da água.
• A produção viral pode interferir na síntese proteica celular e, portanto, levar à morte celular.
• As membranas plasmáticas das células T infectadas por HIV se fundem com células T CD4+ não infectadas em virtude das interações gp120-CD4, e as células gigantes multinucleadas ou sincícios são formadas. O processo de formação do sincício induzido pelo HIV pode ser letal às células T infectadas pelo HIV, bem como para as células T CD4+ que se fundem às células infectadas. No entanto, este fenômeno tem sido amplamente observado in vitro, e os sincícios são raramente vistos nos tecidos de pacientes com AIDS.
Os mecanismos, além de lise direta das células T CD4+ infectadas pelo vírus foram propostos para o esgotamento e perda da função destas células nos indivíduos infectados pelo HIV. Um mecanismo que foi sugerido está relacionado à ativação crônica das células não infectadas pelas infecções que são comuns nos pacientes infectados com o HIV e também pelas citocinas produzidas em resposta a estas infecções. A ativação crônica das células T pode predispor as células a apoptose; a via molecular envolvida neste tipo de morte celular induzida pela ativação ainda não está definida. A morte por apoptose dos linfócitos ativados pode ser responsável pela observação que a perda de células T excede grandemente o número de células infectadas pelo HIV. As CTL específicas do HIV estão presentes em muitos pacientes com AIDS, e estas células podem matar células T CD4+ infectadas. Além disso, os anticorpos contra as proteínas do envelope do HIV podem se ligar às células T CD4+ infectadas por HIV e orientar as células para a citotoxicidade mediada por células dependentes de anticorpos. A ligação da gp120 ao CD4 intracelular recém-sintetizado pode interferir no processamento da proteína normal no retículo endoplasmático e bloquear a expressão da superfície celular do CD4, tornando as células incapazes de responder ao estímulo antigênico. Também foi sugerido que a maturação das células T CD4+ no timo se torna defeituosa nos indivíduos infectados. A importância relativa destes mecanismos indiretos de depleção de células T CD4+ nos pacientes infectados por HIV é incerta e controversa.
Os defeitos funcionais no sistema imunológico dos indivíduos infectados por HIV exacerbam a deficiência imunológica provocada pela depleção de células T CD4+. Estes defeitos funcionais incluem uma redução nas respostas das células T aos antígenos e as respostas imunológicas humorais fracas, embora os níveis séricos totais de Ig possam ser elevados. Os defeitos podem ser um resultado dos efeitos diretos da infecção por HIV nas células T CD4+, incluindo os efeitos da gp120 solúvel liberada das células infectadas ligadas às células não infectadas. Por exemplo, o CD4 que tem gp120 vinculado pode não estar disponível para interagir com as moléculas MHC de classe II nos APCs, e assim as respostas das células T aos antígenos seria inibida. Alternativamente, a ligação de g120 ao CD4 pode emitir sinais de que a função das células T auxiliares T está desregulada. As células T infectadas por HIV são incapazes de formar sinapses justas com as APC, e isto também pode interferir com a ativação das células T. Alguns estudos demonstraram que os pacientes com infecção por HIV têm números elevados de células T CD4+ CD25+ reguladoras, mas ainda não está claro se esta é uma descoberta consistente ou se estas células realmente contribuem para a imunidade defeituosa.
A proteína Tat pode desempenhar algum papel na patogênese da imunodeficiência provocada pelo HIV. Dentro das células T, a Tat pode interagir com uma variedade de proteínas reguladoras, e estas interações podem interferir com as funções da célula T, tais como a síntese das citocinas. Notavelmente, a Tat não só entra no núcleo das células T infectadas, mas também pode escapar através da membrana plasmática e entrar nas células vizinhas, interferindo assim na ativação das células T não infectadas de forma parácrina.
Os macrófagos, as células dendríticas e as células dendríticas foliculares são infectados ou lesionados pelo HIV, e suas anormalidades também contribuem para a progressão da imunodeficiência.
• Os macrófagos expressam níveis muito mais baixos de CD4 do que os linfócitos T auxiliares expressam, mas eles expressam coreceptores CCR5 e são suscetíveis à infecção por HIV. No entanto, os macrófagos são relativamente resistentes aos efeitos citopáticos do HIV. Os macrófagos também podem ser infectados por uma rota independente gp120/gp41, tais como a fagocitose de outras células infectadas ou da endocitose mediada pelo receptor Fc dos virions do HIV revestidos com anticorpos. Como os macrófagos podem ser infectados, mas geralmente não são mortos pelo vírus, eles podem se tornar um reservatório para o vírus. Na verdade, a quantidade de HIV associados aos macrófagos excede os vírus associados à célula T na maioria dos tecidos dos pacientes com AIDS, incluindo o cérebro e o pulmão. Os macrófagos infectados por HIV podem ser prejudicados nas funções de apresentação de antígenos e na secreção de citocinas.
• As células dendríticas também podem ser infectadas pelo HIV. Como os macrófagos, as células dendríticas não são diretamente prejudicadas pela infecção por HIV. No entanto, estas células formam um contanto íntimo com células T puras durante o curso da apresentação dos antígenos. Propõe-se que as células dendríticas infectem as células T virgens (naïve) durante estes encontros, podendo, assim, ser uma importante via para a lesão da célula T.
• As células dendríticas foliculares (FDCs), nos centros germinativos dos linfonodos e nas grandes quantidades de HIV apanhadas pelo baço em suas superfícies, em parte pela ligação dos vírus revestidos de anticorpos mediados pelo receptor Fc. Embora as FDCs não sejam eficientemente infectadas, elas contribuem para a patogênese da imunodeficiência associada ao HIV de pelo menos duas formas. Primeiro, a superfície da FDC é um reservatório para o HIV que pode infectar os macrófagos e as células T CD4+ nos linfonodos. Segundo, as funções normais das FDC nas respostas imunológicas são prejudicadas, e elas podem ser destruídas pelo vírus. Embora os mecanismos da morte induzida pelo HIV das FDCs não sejam compreendidos, o resultado líquido da perda da rede da FDC nos linfonodos e o baço é uma dissolução profunda da arquitetura do sistema linfoide periférico.
Os Reservatórios de HIV e o Volume Viral
O vírus detectado no sangue dos pacientes é produzido principalmente por células T CD4+ infectadas de curta duração e em menor quantidade por outras células infectadas. Três fases de decadência da viremia do plasma foram observadas nos pacientes tratados com medicamentos antirretrovirais ou previstas por modelos matemáticos, e estas curvas de decadência foram usadas para considerar a distribuição do HIV nos diferentes reservatórios celulares. Acredita-se que mais de 90% do vírus do plasma seja produzido por células de curta duração (meia-vida de um dia), que são provavelmente ativadas por células T CD4+ que são grandes reservatórios e fontes do vírus em pacientes infectados. Cerca de 5% do vírus do plasma é produzido por macrófagos, que têm um volume mais lento (meia-vida de cerca de 2 semanas). A hipótese é que uma pequena fração do vírus, talvez tão pouco como 1%, esteja presente nas células de memória T infectadas de forma latente. Devido à longa vida útil das células de memória, poderia levar décadas para este reservatório do vírus ser eliminado, mesmo se todos os novos ciclos de infecção fossem bloqueados.
Características Clínicas do HIV
Uma vasta quantia de informações se acumulou sobre a epidemiologia e a evolução clínica da infecção por HIV. Como a terapia do medicamento antirretroviral está melhorando, muitas das manifestações estão mudando. Na seção seguinte, descrevemos as características “clássicas” da infecção por HIV e chamamos a atenção para as imagens da mudança quando relevante.
Transcrição do HIV e Epidemiologia da AIDS
O HIV é transmitido de um indivíduo para outro por três vias principais:
• O contato sexual é o modo mais frequente de transmissão, tanto entre casais heterossexuais (o modo mais frequente de transmissão na África e na Ásia) ou entre parceiros masculinos homossexuais. Na África subsaariana, onde a taxa de infecção é a mais alta do mundo (estimada em cerca de 10.000 novos casos todos os dias), mais da metade dos indivíduos infectados são mulheres.
• A transmissão de mãe para filho do HIV representa a maioria dos casos pediátricos de AIDS. Este tipo de transmissão ocorre mais frequentemente no útero ou durante o parto, embora a transmissão através do leite materno também seja possível.
• A inoculação de um recebedor com produtos de sangue infectado ou sangue também é um modo frequente de transmissão de HIV. Agulhas compartilhadas por usuários de drogas intravenosas são responsáveis pela maioria dos casos desta forma de transmissão. Com o advento da triagem laboratorial de rotina, a transfusão de sangue ou os hemoderivados em um ambiente clínico são responsáveis por uma pequena parcela das infecções por HIV.
Curso Clínico de Infecção por HIV
A evolução das doenças provocadas por HIV pode ser seguida medindo a quantidade de vírus no plasma do paciente e pela contagem de células sanguíneas T CD4+ (Fig. 20-8).
• A fase aguda da doença, também chamada de síndrome do HIV agudo, é o período da viremia caracterizada por sintomas inespecíficos da infecção. Ela se desenvolve em 50% a 70% dos adultos infectados, normalmente de 3 a 6 semanas após a infecção. Há um aumento do vírus do plasma e uma modesta redução na contagem de células T CD4+, mas o número de células sanguíneas T CD4+ geralmente retorna ao normal. Em muitos pacientes, no entanto, a infecção é oculta e não há sintomas.
• A fase crônica da latência clínica pode durar por muitos anos. Durante este tempo, o vírus está contido dentro dos tecidos linfoides, e a perda de células T CD4+ é corrigida pela reposição de progenitores. Os pacientes são assintomáticos ou sofrem de infecções secundárias. Dentro de 2 a 6 meses após a infecção, a concentração do vírus do plasma se estabiliza em um determinado ponto de definição, que difere entre os pacientes. O nível do ponto de definição viral e o número de células sanguíneas T CD4+ são indicadores clinicamente úteis da progressão da doença. À medida que a doença progride, os pacientes se tornam suscetíveis a outras infecções, e as respostas imunológicas a estas infecções podem estimular a produção de HIV e acelerar a destruição dos tecidos linfoides. Conforme discutido anteriormente, a transcrição genética do HIV pode ser reforçada por estímulos que ativam as células T, tais como os antígenos e uma variedade de citocinas. As citocinas, como o TNF, que são produzidas pelo sistema imunológico inato em resposta às infecções microbianas, são particularmente eficazes no aumento da produção do HIV. Assim, à medida que o sistema imunológico tenta erradicar outros micro-organismos, ele traz sua própria destruição pelo HIV.
• O HIV progride para a fase final e quase invariavelmente letal, chamada AIDS, quando a contagem de células sanguíneas T CD4+ cai para menos de 200 células/mm3. A viremia do HIV pode subir drasticamente à medida que a replicação viral nos reservatórios e não nas células T acelere de forma não verificada. Os pacientes com AIDS sofrem de combinações de infecções oportunistas, neoplasmos, caquexias (síndrome de perda de peso no HIV), insuficiência renal (nefropatia de HIV) e degeneração do SNC (encefalopatia da AIDS) (Tabela 20-7). Como os CD4+ as células T auxiliares são essenciais tanto para as respostas imunes mediadas por células como humorais para vários micro-organismos, a perda destes linfócitos é a principal razão pela qual os pacientes com AIDS se tornam suscetíveis a diversos tipos de infecções. Além disso, muitos dos tumores que surgem em pacientes com AIDS têm etiologia viral, e sua prevalência no cenário da AIDS reflete a incapacidade do paciente infectado por AIDS de estabelecer uma resposta imune eficaz contra os vírus oncogênicos. A caquexia é frequentemente vista em pacientes com doenças inflamatórias crônicas e pode ser resultado dos efeitos das citocinas inflamatórias (como a TNF) sobre o apetite e o metabolismo. A doença do SNC na AIDS pode ser devida à lesão neuronal pelo vírus ou por lançar proteínas virais, como a gp120 e a Tat, bem como os efeitos das citocinas elaboradas pelas células microgliais infectadas. Muitas destas consequências devastadoras da infecção por HIV, incluindo as infecções oportunistas e os tumores, foram significantemente reduzidas pela terapia antirretroviral altamente ativa.
TABELA 20-7 Características Clínicas da Infecção por HIV
| Fase da Doença | Característica Clínica |
|---|---|
| Doença aguda pelo HIV | Febre, dores de cabeça, dor de garganta com faringite, linfadenopatia generalizada, erupções |
| Período de latência clínica | Diminuição da contagem de células T CD4+ sanguíneas |
| AIDS |
Embora este resumo do curso clínico seja válido para os casos mais graves, a taxa de progressão da doença é muito variável, e alguns indivíduos não são progressores em longo prazo. Os correlatos imunológicos de progressão variável permanecem desconhecidos. Além disso, a recente terapia antirretroviral mudou o curso da doença e reduziu consideravelmente a incidência de infecções oportunistas graves (Pneumocystis) e tumores (como o sarcoma de Kaposi).
Respostas Imunes ao HIV
As respostas imunes mediadas por células e humorais específicos do HIV se desenvolvem após a infecção, mas geralmente oferecem uma proteção limitada. A resposta inicial à infecção por HIV é, na verdade, semelhante em muitos aspectos à resposta imune a outros vírus e serve para limpar a maioria dos vírus presentes no sangue e na circulação das células T. No entanto, é claro que estas respostas imunes falham em erradicar todos os vírus e a infecção eventualmente sobrecarrega o sistema imunológico na maioria dos indivíduos. Apesar da falta de eficácia das respostas imunes ao vírus, é importante caracterizá-las por três razões. Primeiro, a resposta imune pode ser prejudicial ao hospedeiro, por exemplo, estimulando a captação do vírus opsonizado nas células não infectadas pela endocitose mediada pelo receptor Fc ou pela erradicação das células T CD4+ expressando antígenos virais através dos CTL CD8+. Segundo, os anticorpos contra o HIV são marcadores de diagnósticos da infecção por HIV, que são amplamente usados para fins de triagem. Terceiro, o design de vacinas eficazes para a imunização contra o HIV requer conhecimento dos tipos de respostas imunes mais prováveis de serem protetoras (“as correlatas de proteção”).
Muitas respostas imunes natas contra o HIV foram descritas. Estas incluem os peptídeos antimicrobianos (desensinas), células NK, células dendríticas (especialmente as células dendríticas plasmacitoides produzindo interferons tipo I) e o sistema complementar. O papel destas respostas no combate à infecção não é estabelecido.
A resposta imune adaptativa inicial à infecção por HIV é caracterizada pela expansão das células T CD8+ específicas para peptídeos de HIV. Até 10% ou mais das células em circulação T CD8+ podem ser específicos para o HIV durante os estágios iniciais da infecção. Estes CTL controlam a infecção na fase aguda (Fig. 20-8), mas em última análise se revelam ineficazes devido ao surgimento de mutantes de escape viral (variantes com antígenos modificados). Células T CD4+ também respondem ao vírus, e estas células T CD4+ podem contribuir para o controle viral de diversas maneiras. Uma resposta eficaz da célula T CD4+ é necessária como fonte de auxílio para a geração de células T CD8+ de memória, mas as células T CD4+ também mostraram mediar respostas citolíticas contra as células infectadas por HIV, talvez usando ligante Fas para atingir o Fas nas células T CD4+ infectadas.
A importância das respostas da CTL no controle do HIV é ressaltada pela evolução do vírus sob a pressão imunitária, resultando no isolamento viral que perdeu seus epítopos dos CTL originais. A evolução do vírus também resulta na perda dos epítopos reconhecidos pelas células T CD4+, indicando que tanto as células CD8+ como as células CD4+ contribuem para a defesa do hospedeiro contra o vírus.
As respostas dos anticorpos a uma variedade de antígenos do HIV são detectáveis dentro de 6 a 9 semanas após a infecção. As moléculas mais imunogênicas do HIV que provocam respostas dos anticorpos parecem ser as glicoproteínas do envelope, e os altos títulos dos anticorpos da anti-gp120 e da anti-gp41 estão presentes na maioria dos indivíduos infectados pelo HIV. Outros anticorpos do anti-HIV encontrados frequentemente nos soros dos pacientes incluem anticorpos para a transcriptase reversa p24, e os produtos gag e pol (Fig. 20-8). Os efeitos destes anticorpos no curso clínico da infecção por HIV são incertos. Os anticorpos iniciais não são neutralizantes e geralmente não são bons inibidores de infecção viral ou de efeitos citopáticos. Os anticorpos neutralizantes contra a gp120 se desenvolvem de 2 a 3 meses após a infecção primária, mas mesmo estes anticorpos não conseguem lidar com um vírus que é capaz de mudar rapidamente os epítopos imunodominantes das glicoproteínas de seu envelope.
Mecanismos de Evasão Imunológica pelo HIV
O HIV é o protótipo de um patógeno infeccioso que evade as defesas do hospedeiro destruindo o sistema imune. Além disso, diversas características do HIV podem auxiliar o vírus a evadir a imunidade do hospedeiro.
O HIV tem uma taxa de mutação extremamente alta devido a uma transcrição reversa sujeita a falhas, e desta forma pode evadir a detecção através dos anticorpos ou células T geradas em resposta às proteínas virais. Estima-se que em uma pessoa infectada, cada possível ponto de mutação no genoma viral ocorre todos os dias. A região da molécula gp120, chamada de circuito V3, é um dos componentes mais antigenicamente variáveis; ele varia no isolamento do HIV retirado do mesmo indivíduo em momentos diferentes. Além disso, as regiões do circuito V3 que são críticos para a entrada viral e, portanto, são menos frequentemente modificados, não são facilmente expostas ao sistema imune humoral.
As células infectadas pelo HIV podem evadir os CTLs através do desequilíbrio da expressão da molécula MHC de classe I. A proteína Nef do HIV inibe a expressão das moléculas MHC de classe I, principalmente através da promoção da internalização destas moléculas. Outros mecanismos de inibição da imunidade mediada por células foram demonstradas em alguns casos. Estes incluem uma inibição preferencial das citocinas TH1, ativação das células T reguladoras e supressão das funções das células dendríticas. Os mecanismos destas ações do vírus, bem como seu significado patogênico não são estabelecidos.
Controladores de Elite e não Progressores a Longo Prazo: Um Possível Papel para os Genes Hospedeiros
Embora a maioria dos indivíduos infectados com o HIV por fim desenvolverá a AIDS, cerca de 1% dos indivíduos infectados não desenvolvem a doença. Estes indivíduos têm alta contagem de células T CD4+ e CD8+, não precisam de terapia, e têm viremia persistente, mas nenhuma doença por pelo menos 10 a 15 anos. Com base no grau de viremia, este grupo pode ser divido em dois subgrupos: os não progressores em longo prazo têm viremia detectável de cerca de 5.000 cópias de HIV-1 RNA por mililitro de sangue; e um subgrupo muito menor de “controladores de elite” apresenta cargas virais de cerca de 50 cópias ou menos de HIV-1 RNA por mililitro de sangue. Há um considerável interesse na compreensão da base genética do controle do HIV através do exame destes grupos de indivíduos em detalhes. Até agora, um forte papel para o lócus MHC na proteção de indivíduos e na prevenção da progressão foi sugerido por estudos de associação genética. HLA específicos de classe I e até mesmo alguns loci HLA de classe II foram ligados à ausência da progressão da doença. Mencionamos anteriormente a importância da herança da deleção de 32-bp homozigóticos CCR5 na proteção contra infecções, e outros fatores genéticos que contribuem para a resistência provavelmente serão revelados nos próximos anos.
Tratamento e Prevenção da AIDS e Desenvolvimento de Vacinas
Os esforços das pesquisas ativas foram destinados a desenvolver reagentes que interferem no ciclo de vida viral. O tratamento da infecção por HIV e por AIDS agora inclui a administração de três classes de medicamentos antivirais, usados em combinação, que visam moléculas virais para as quais nenhum homólogo humano existe. Os primeiros medicamentos antirretrovirais a serem amplamente utilizados foram análogos de nucleosídeos que inibem a atividade da transcriptase reversa. Estes medicamentos incluem os análogos de nucleosídeos de deoxitimidina, tais como o 3′-azido-3′-deoxitimidina (AZT), análogos de nucleosídeos de deoxicitina e os análogos de deoxiadenosina. Quando estes medicamentos são utilizados sozinhos, eles são geralmente eficazes na redução significativa dos níveis de HIV RNA durante diversos meses a anos, mas eles geralmente não detêm a progressão da doença induzida pelo HIV, principalmente devido à evolução do vírus com formas modificadas de transcriptase reversa que são resistentes aos medicamentos. Os inibidores não nucleosídeos de transcriptase reversa se ligam diretamente à enzima e inibem sua função. Foram desenvolvidos inibidores da protease viral que bloqueiam o processamento das proteínas precursoras em capsídeos virais maduros e proteínas essenciais. Quando estes inibidores de protease são usados sozinhos, os vírus mutantes resistentes a seus efeitos rapidamente emergem. No entanto, os inibidores de protease agora estão sendo usados em combinação com dois inibidores diferentes de transcriptase reversa. Esta nova terapia com três medicamentos, comumente referida como HAART (terapia antirretroviral altamente ativa) ou ART (terapia antirretroviral) provou ser eficaz na redução do RNA viral do plasma para níveis indetectáveis na maioria dos pacientes tratados por até 3 anos. Um inibidor da integrase agora também é usado como parte da terapia antiviral. Os “inibidores de entrada”, que impedem a entrada viral, visando ou o CD4 ou o CCR5 na célula do hospedeiro ou a gp41 ou a gp120 no vírus, são outra nova categoria da terapêutica. O Maraviroc é um inibidor de entrada que bloqueia o CCR5 e, assim, impede a entrada viral. Os alimentos que tem o gp41 como alvo incluem compostos como a enfuvirtida que impedem a fusão do envelope viral com a membrana plasmática da célula hospedeira. Embora a terapia antirretroviral tenha reduzido os títulos virais para baixo do limite de detecção em até 10 anos em alguns pacientes, é pouco provável que este tratamento possa eliminar o vírus de todos os reservatórios (especialmente as células infectadas a um longo tempo), e a resistência aos medicamentos pode vir a se desenvolver. Outros problemas formidáveis associados a estas terapias com novos medicamentos, que prejudicarão seu uso eficaz em muitas partes do mundo, incluem o gasto elevado, horários de administração complicados e efeitos adversos significativos.
As infecções individuais vividas por pacientes com AIDS são tratadas com a profilaxia adequada, antibióticos e medidas de suporte. A terapia com antibióticos mais agressivos é muitas vezes necessária do que para infecções semelhantes em hospedeiros menos comprometidos.
Os esforços na prevenção da infecção por HIV são extremamente importantes e potencialmente eficazes no controle da epidemia por HIV. Nos Estados Unidos, a triagem da rotina dos produtos sanguíneos para a evidência da infecção por HIV do doador já reduziu o risco deste modo de transmissão a níveis significantes. Várias medidas de saúde pública para aumentar o uso de preservativos e reduzir o uso de agulhas contaminadas por usuários de drogas intravenosas estão agora disseminadas. Talvez os esforços mais eficazes na prevenção sejam as campanhas para aumentar a consciência pública sobre o HIV.
O desenvolvimento de uma vacina eficaz contra o HIV é uma prioridade para as instituições de pesquisa biomédica em todo o mundo. A tarefa tem sido dificultada pela capacidade do vírus de se modificar e variam muito de seus antígenos imunogênicos. É provável que uma vacina eficaz tenha que estimular tanto a resposta humoral como a mediada por células aos antígenos virais que são críticas para o ciclo de vida viral. Para atingir este objetivo, diversas abordagens estão sendo tentadas para o desenvolvimento de vacinas contra o HIV. Grande parte do trabalho preliminar envolveu a infecção pelo vírus de imunodeficiência símia (SIV) dos macacos, e vacinas eficazes contra o SIV já foram desenvolvidas. Este sucesso é animador por que o SIV está molecularmente relacionado muito próximo ao HIV e provoca uma doença nos macacos que é semelhante à AIDS nos humanos. Várias vacinas do vírus vivo foram testadas na esperança de que elas induzam fortes respostas à CTL. Estas vacinas incluem vírus híbridos recombinantes não virulentos compostos em parte de sequências de SIV e em parte de sequências de HIV ou vírus que foram atenuados pelas exclusões em uma ou mais partes dos genomas virais, como o gene nef. Uma preocupação com as vacinas dos vírus vivos é o seu potencial para provocar doenças se não forem completamente atenuados e, possivelmente, recombinar-se com o tipo selvagem de HIV para produzir uma variante patogênica. Outra abordagem que evita este problema de segurança, mas mantém a eficácia na redução da imunidade mediada pela CTL é o uso de vetores virais dissociados do HIV recombinantes vivos que transportam genes do HIV. Testes preliminares em voluntários humanos já mostraram que as vacinas canarypox que expressam diversos genes HIV-1 podem induzir fortes respostas da CTL aos antígenos do HIV. Muitas vacinas de DNA também foram estudadas; estas vacinas são compostas de combinações de genes estruturais e reguladores do SIV e do HIV embalados em vetores de expressão de DNA de mamíferos. As combinações de vacinas, tais como a imunização inicial com uma vacina de DNA seguida de reforço com um vetor canarypox que expressam os genes do HIV, produziram alguns dos mais promissores resultados até o momento. A proteína recombinante ou vacinas de subunidades de peptídeos que provocam os anticorpos têm sido até agora de valor limitado porque os anticorpos induzidos por estas vacinas tipicamente não neutralizam isolamentos clínicos de HIV. As tentativas estão atualmente em curso para gerar vacinas que podem induzir a neutralização de anticorpos contra o HIV.
• As doenças de imunodeficiência são provocadas por defeitos congênitos ou adquiridos nos linfócitos, fagócitos e outros mediadores da imunidade adaptativa e inata. Estas doenças são associadas a um aumento da suscetibilidade à infecção, a natureza e gravidade dos quais dependem em grande parte sobre que componente do sistema imune é anormal e a extensão da anormalidade.
• Os distúrbios da imunidade inata incluem os defeitos na morte microbial pelos fagócitos (p. ex., a CGD ou síndrome de Chédiak-Higashi), a migração e adesão de leucócitos (p. ex., a deficiência de adesão leucocitária), a sinalização do TLR e os complementos (Cap. 12).
• As imunodeficiências combinadas graves incluem defeitos no desenvolvimento de linfócitos que afetam tanto as células T como as B e são provocados pela sinalização defeituosa da citocina, metabolismo anormal das purinas, recombinação defeituosa do V(D)J, e mutações que afetam a maturação das células T.
• As imunodeficiências dos anticorpos incluem doenças provocadas pela maturação defeituosa das células B ou da ativação e dos defeitos na colaboração das células T-B (síndrome do hiper-IgM ligado ao X).
• As imunodeficiências das células T incluem as doenças em que a expressão das moléculas MHC é defeituosa, células T que sinalizam distúrbios, e doenças raras que envolvem funções das células CTL e assassinas naturais (natural killer – NK).
• O tratamento das imunodeficiências congênitas envolve transfusões de anticorpos, transplante de medula óssea ou de célula-tronco, ou substituição enzimática. A terapia genética pode oferecer tratamentos aprimorados no futuro.
• As imunodeficiências adquiridas são provocadas por infecções, desnutrição, câncer disseminado, e terapia imunossupressora para rejeição de transplante ou doenças autoimunes.
• A AIDS é uma imunodeficiência grave provocada por infecção por HIV. Este vírus RNA infecta os linfócitos T CD4+, macrófagos, e células dendríticas e provoca disfunção progressiva do sistema imunitário. A maioria da imunodeficiência na AIDS pode ser atribuída ao esgotamento das células T CD4+.
• O HIV entra nas células através ligando-se a ambas as moléculas CD4 e a um coreceptor da família de receptores de quimiocina. Uma vez que está dentro da célula, o genoma viral é reversamente transcrito no DNA e incorporado no genoma celular. A transcrição dos genes virais e a reprodução viral são estimulados por sinais que normalmente ativam a célula hospedeira. A produção do vírus é acompanhada pela morte das células infectadas.
• A fase aguda da infecção é caracterizada pela morte das células de memória T CD4+ nos tecidos da mucosa e na disseminação do vírus para os linfonodos. Na fase latente subsequente, há baixo nível de replicação de vírus nos tecidos linfoides e perda lenta e progressiva das células T. A ativação persistente das células T promove sua morte, levando à perda rápida e à deficiência imunológica na fase crônica da infecção.
• A depleção das células T CD4+ em indivíduos infectados pelo HIV se deve aos efeitos citopáticos diretos do vírus, efeitos tóxicos dos produtos virais, tais como o abrigo de gp120, e os efeitos indiretos, tais como a morte celular induzida por ativação ou a morte da CTL ou células CD4+ infectadas.
• Diversos reservatórios de HIV existem nos indivíduos infectados, incluindo células T CD4+ ativadas transitórias, macrófagos de vida mais longa, e células T de memória infectadas de forma latente com vida muito longa.
• A depleção das células T CD4+ induzida pelo HIV resulta na redução da suscetibilidade à infecção por uma série de micro-organismos oportunistas. Além disso, os pacientes infectados pelo HIV têm aumento de incidência de tumores, especialmente o sarcoma de Kaposi e os linfomas das células B associados ao EBV, e a encefalopatia. A incidência destas complicações tem sido muito reduzida pela terapia antirretroviral.
• O HIV tem uma alta taxa de mutação, que permite que o vírus evada as respostas imunológicas do hospedeiro e se torne resistente às terapias medicamentosas. A variabilidade genética também representa um problema para a concepção de uma vacina eficaz contra o HIV. A infecção por HIV pode ser tratada por uma combinação de inibidores de enzimas virais.
Imunodeficiências Congênitas (Primárias)
Blackburn MR, Kellems RE. Adenosine deaminase deficiency: metabolic basis of immune deficiency and pulmonary inflammation. Advances in Immunology. 2005;86:1-4.
Bonilla FA, Geha RS. Update on primary immunodeficiency diseases. Journal of Allergy and Clinical Immunology. 2006;117(Suppl):435-441.
Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annual Review of Immunology. 2004;22:625-655.
Bustamante J, Boisson-Dupuis S, Jouanguy E, Picard C, Puel A, Abel L, Casanova JL. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases. Current Opinion in Immunology. 2008;20:39-48.
Castigli E, Geha RS. Molecular basis of common variable immunodeficiency. Journal of Allergy and Clinical Immunology. 2006;117:740-746.
Chen X, Jensen PE. MHC class II antigen presentation and immunological abnormalities due to deficiency of MHC class II and its associated genes. Experimental and Molecular Pathology. 2008;85:40-44.
Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, Coustan-Smith E, Howard V, Campana D. Primary B cell immunodeficiencies: comparisons and contrasts. Annual Review of Immunology. 2009;27:199-227.
Cunningham-Rundles C, Ponda PP. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nature Reviews Immunology. 2005;5:880-892.
Fischer A. Human primary immunodeficiency diseases. Immunity. 2008;28:835-846.
Groneberg DA, Quarcoo D, Frossard N, Fischer A. Gene therapy for severe combined immunodeficiency. Annual Review of Medicine. 2005;56:585-602.
Janka GE. Hemophagocytic lymphohistiocytosis. Hematology. 2005;10(Suppl 1):104-107.
Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nature Reviews Molecular and Cell Biology. 2008;9:759-769.
Notarangelo LD. Primary immunodeficiencies. Journal of Allergy and Clinical Immunology. 2010;125:S182-S194.
Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. Journal of Allergy and Clinical Immunology. 2006;117:725-738.
Puel A, Yang K, Ku CL, et al. Heritable defects of the human TLR signaling pathways. Journal of Endotoxin Research. 2005;11:220-224.
Barouch DH. Challenges in the development of an HIV-1 vaccine. Nature. 2008;455:613-619.
Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nature Immunology. 2006;7:235-239.
Derdeyn CA, Silvestri G. Viral and host factors in the pathogenesis of HIV infection. Current Opinion in Immunology. 2005;17:366-373.
Haase AT. Perils at mucosal front lines for HIV and SIV and their hosts. Nature Reviews Immunology. 2005;5:783-792.
Hladik F, McElrath MJ. Setting the stage: host invasion by HIV. Nature Reviews Immunology. 2008;8:447-457.
Johnston MI, Fauci AS. An HIV vaccine—evolving concepts. New England Journal of Medicine. 2007;356:2073-2081.
Letvin NL. Progress and obstacles in the development of an AIDS vaccine. Nature Reviews Immunology. 2006;6:930-939.
Lusso P. HIV and the chemokine system: 10 years later. EMBO Journal. 2006;25:447-456.
Mascola JR, Montefiori DC. The role of antibodies in HIV vaccines. Annual Review of Immunology. 2010;28:413-444.
McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nature Reviews Immunology. 2010;10:11-23.
Nixon DF, Aandahl EM, Michaelsson J. CD4+CD25+ regulatory T cells in HIV infection. Microbes and Infection. 2005;7:1063-1065.
Walker BD, Burton DR. Towards an AIDS vaccine. Science. 2008;320:760-764.