CAPÍTULO 14 Tolerância Imunológica e Autoimunidade
A tolerância imunológica é definida como a não responsividade a um antígeno, induzida pela exposição prévia a este antígeno. Quando linfócitos específicos encontram antígenos, o linfócito pode ser ativado, iniciando uma resposta imunológica a este antígeno, ou essas células podem ficar inativas ou ser eliminadas, levando à tolerância. Diferentes formas do mesmo antígeno podem induzir uma resposta imunológica ou tolerância. Antígenos que têm a capacidade de induzir tolerância são chamados de tolerógenos, ou antígenos tolerogênicos, para distingui-los dos imunógenos, que geram imunidade. Um único antígeno pode ser um imunógeno ou um tolerógeno, dependendo das condições em que é exposto a linfócitos específicos (p. ex., na presença ou ausência, respectivamente, de inflamação e respostas imunes inatas). A tolerância a autoantígenos, também chamada de autotolerância, é uma propriedade fundamental do sistema imune normal, e uma falha na autotolerância resulta em reações imunes contra antígenos próprios (autólogos). Esse tipo de reação é chamado de autoimunidade, e as doenças que ela pode causar são conhecidas como doenças autoimunes. A elucidação dos mecanismos de autotolerância é a chave para o entendimento da patogênese da autoimunidade.
Neste capítulo, discutiremos a tolerância imunológica principalmente no contexto da autotolerância e como a autotolerância pode falhar, resultando na autoimunidade. Mencionaremos também a relevância da tolerância a não responsividade a antígenos estranhos e o potencial de indução de tolerância como estratégia terapêutica para doenças imunológicas e para prevenir a rejeição de transplantes de células e órgãos. Devido à importância da autotolerância para a saúde dos indivíduos e à promessa terapêutica da tolerância, há grande interesse em compreender esse fenômeno e aprender como aplicá-lo a humanos.
CARACTERÍSTICAS GERAIS DA TOLERÂNCIA IMUNOLÓGICA
Há diversas características de tolerância em populações de linfócitos T e B. É importante entender os princípios gerais antes de discutirmos os mecanismos específicos de tolerância nesses linfócitos.
• Indivíduos normais são tolerantes aos seus antígenos (antígenos próprios) porque os linfócitos que reconhecem antígenos próprios são destruídos ou inativados, ou porque a especificidade desses linfócitos é alterada. Todos os indivíduos herdam essencialmente os mesmos segmentos de genes para receptores de antígenos, e estes genes se recombinam e são expressos nos linfócitos após a sua diferenciação das células-tronco. As especificidades dos receptores codificadas pelos genes recombinados são aleatórias, e não são influenciadas pelo que é estranho ou próprio de cada indivíduo (Cap. 8). Não é de surpreender que durante este processo de gerar um grande e variado repertório, algumas células T e B em desenvolvimento em cada indivíduo possam expressar receptores capazes de reconhecer moléculas normais nesse indivíduo (i. e., antígenos próprios). Portanto, há o risco dos linfócitos reagirem contra as células e tecidos deste indivíduo, causando doenças. Os mecanismos de tolerância imunológica são criados para prevenir tais reações.
A importância da autotolerância para a saúde dos indivíduos foi considerada fundamental desde os primórdios da imunologia. No Capítulo 1, introduzimos o conceito de discriminação do não próprio, que é a habilidade do sistema imunológico de reconhecer e responder aos antígenos estranhos, mas não aos antígenos próprios. Macfarlane Burnet acrescentou à sua hipótese da seleção clonal o corolário que linfócitos específicos para os antígenos próprios são eliminados a fim de evitar uma reação imunológica contra os tecidos do próprio indivíduo. No final deste capítulo, veremos que a autotolerância é mantida por vários diferentes mecanismos que previnem a maturação e a ativação de linfócitos potencialmente autorreativos.
• A tolerância é resultante do reconhecimento do antígeno por linfócitos específicos. Em outras palavras, a tolerância, em sua definição plena, é antígeno específico. Isso contrasta com a imunossupressão terapêutica e com as imunodeficiências herdadas ou adquiridas, que afetam linfócitos de muitas especificidades. Os avanços fundamentais que permitiram aos imunologistas estudarem a tolerância foram a indução deste fenômeno em animais pela exposição a antígenos definidos, sob várias condições, e, mais tarde, a análise das funções dos linfócitos que tinham encontrado antígenos tolerogênicos. Os resultados que estabelecem definitivamente a tolerância como um fenômeno imunologicamente específico que pode ser induzido por experimentação originam-se de estudos de rejeição de enxertos em camundongos endogâmicos realizados por Peter Medawar e colegas nos anos de 1950. Um animal adulto de uma linhagem A rejeita um enxerto de pele de um camundongo alogênico de uma linhagem B por diferir da linhagem A no complexo principal de histocompatibilidade (MHC). Se forem injetados glóbulos brancos do camundongo da linhagem B em um camundongo da linhagem A durante a fase neonatal (onde as células servem como fonte de antígenos da linhagem B), essas células não serão rejeitadas (porque o camundongo neonato é imunodeficiente), e um pequeno número delas sobreviverá indefinidamente no animal receptor. A persistência de células alogênicas linfoides em um hospedeiro é chamada de microquimerismo hematopoiético. A linhagem A receptora passará a aceitar o enxerto da linhagem B mesmo quando se tornar adulta. No entanto, a linhagem A receptora rejeitará qualquer enxerto de pele de todas as linhagens de camundongos que tenham MHC diferentes da linhagem B. Assim, a tolerância aos enxertos é imunologicamente específica. Esses experimentos levam ao conceito de que a exposição de antígenos estranhos aos linfócitos em desenvolvimento induz tolerância a estes antígenos. O microquimerismo está sendo estudado como uma possível abordagem para a prevenção de rejeição de enxertos em humanos (Cap. 16).
• A autotolerância pode ser induzida em linfócitos autorreativos imaturos nos órgãos linfóides primários (tolerância central) ou em linfócitos maduros em locais periféricos (tolerância periférica) (Fig. 14-1). A tolerância central assegura que o repertório de linfócitos maduros torna-se incapaz de responder aos antígenos próprios expressos nos órgãos linfoides primários (o timo para as células T e a medula óssea para os linfócitos B, também chamados de órgãos linfoides centrais). No entanto, a tolerância central não é perfeita, e não explica por que o sistema imunológico não responde aos antígenos que estão presentes somente nos tecidos periféricos. A tolerância aos antígenos que são específicos para os tecidos é mantida pelos mecanismos periféricos. Mecanismos adicionais de tolerância periférica funcionam em tecidos periféricos para prevenir a ativação de linfócitos autorreativos que tenham escapado da tolerância central.
• A tolerância ocorre durante a maturação dos linfócitos nos órgãos linfoides centrais (primários), onde todos os linfócitos em desenvolvimento passam por um estágio no qual um encontro com um antígeno pode levar à morte da célula ou à substituição de um antígeno receptor autorreativo por um novo antígeno receptor autorreativo. Os órgãos linfoides primários contêm majoritariamente antígenos próprios e não antígenos estranhos porque estes antígenos estranhos (p. ex., microbianos) que entram do ambiente externo são capturados e transportados para órgãos linfoides periféricos, como os linfonodos, baço e tecidos linfóides mucosos, e não são transportados para o timo ou para a medula óssea. Os antígenos normalmente presentes no timo e na medula óssea incluem antígenos próprios ubíquos, ou amplamente disseminados, inclusive aqueles trazidos pelo sangue. Além disso, alguns antígenos periféricos tecido-específicos são expressos em células especializadas no timo. Portanto, nos órgãos linfóides primários, os linfócitos imaturos que reconhecem os antígenos especificamente são normalmente células específicas para antígenos próprios, e não para antígenos estranhos. O reconhecimento dos antígenos próprios por linfócitos imaturos pode ter diversos resultados: as células podem morrer de apoptose (chamada de deleção clonal ou seleção negativa porque este processo seleciona clones de células antígeno-específicas para eliminação); muitas células B imaturas não morrem, mas mudam seus receptores (chamado de edição de receptor) e, portanto não reconhecem mais o antígeno próprio que disparou este processo; e algumas células T CD4+ se diferenciam em células T reguladoras, que migram para a periferia e impedem respostas aos antígenos próprios (Fig. 14-1).
• A tolerância periférica ocorre quando, em consequência do reconhecimento dos antígenos próprios, os linfócitos maduros tornam-se incapazes de responder a esses antígenos, ou são induzidos a morrer por apoptose, ou células T maduras são ativamente suprimidas por células T reguladoras. A tolerância periférica e muito importante para a manutenção da não responsividade aos antígenos próprios que estão expressos nos tecidos periféricos, e não nos órgãos linfoides primários, e para tolerância aos antígenos próprios que são expressos somente na vida adulta, depois que linfócitos maduros foram gerados. Mecanismos periféricos também podem servir como um back-up para os mecanismos centrais, que podem não eliminar todos os linfócitos autorreativos.
• Se o reconhecimento dos antígenos por linfócitos fica ativado ou tolerante é determinado pelas propriedades dos antígenos, pelo estado de maturação dos linfócitos antígeno específicos, e pelos tipos de estímulos recebidos quando estes linfócitos encontram antígenos próprios. Como veremos neste capítulo, esses fatores afetam o destino dos linfócitos que encontram seus antígenos cognatos de maneiras diferentes.
• Alguns antígenos próprios podem ser ignorados pelo sistema imunológico. A importância deste fenômeno de “ignorância” para a manutenção da autotolerância não foi estabelecida. Alguns antígenos podem ser anatomicamente isolados do sistema imunológico e, portanto não podem engajar receptores de antígeno. Em modelos experimentais, alguns antígenos próprios são reconhecidos por linfócitos, mas, por motivos desconhecidos, deixam de receber qualquer resposta e são funcionalmente ignorados.
• Na ausência de sinais coestimulantes, antígenos estranhos podem inibir a resposta imunológica pela indução de tolerância em linfócitos específicos. Muitos dos mecanismos de tolerância aos antígenos estranhos são similares aos da autotolerância nos linfócitos maduros. Métodos efetivos de imunização são utilizados para aumentar a imunogenicidade dos antígenos para que possam ser administrados por determinadas vias, promovendo a ativação linfocitária e prevenindo a indução de tolerância. Alguns micro-organismos e tumores também podem evadir-se ao ataque imune induzindo falta de responsividade em linfócitos específicos.
• A indução de tolerância imunológica pode ser explorada como uma abordagem terapêutica para prevenir uma resposta imunológica indesejável. Um grande esforço tem sido feito no desenvolvimento de estratégias para a indução de tolerância a fim de prevenir a rejeição de órgãos, e também para o tratamento de doenças alérgicas e autoimunes. A indução de tolerância pode ser útil para a prevenção de reações imunológicas contra novos produtos gênicos utilizados nos protocolos de terapia gênica e, ainda, para a prevenção de reações a proteínas injetadas em pacientes com deficiência de certas proteínas (p. ex., tratamento da hemofilia com o fator VIII) e para a promoção da aceitação de transplantes de células-tronco.
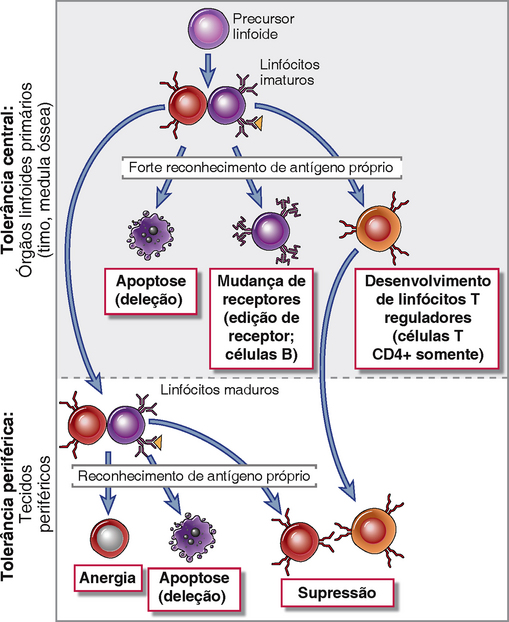
FIGURA 14-1 Tolerância central e periférica a antígenos próprios. Linfócitos imaturos específicos para antígenos próprios podem encontrar estes antígenos nos órgãos linfoides primários e são eliminados, alteram sua especificidade (somente células B), ou (no caso de células T CD4+) se desenvolvem como linfócitos reguladores (tolerância central). Alguns linfócitos autorreativos podem maturar e entrar em tecidos periféricos e podem ser inativados ou eliminados pelo encontro com antígenos próprios nestes tecidos ou ser suprimidos pelas células T reguladoras (tolerância periférica). (Note que as células T reconhecem antígenos apresentados por células apresentadoras de antígenos, que não são mostradas.)
Não sabemos quais antígenos próprios induzem a tolerância central ou periférica (ou são ignorados). É tecnicamente difícil identificar células raras que podem ser específicas para antígenos próprios naturais porque os reagentes para a detecção de linfócitos antígeno específicos não são amplamente usados e poucos antígenos próprios são definidos para os quais tais reagentes poderiam ser produzidos. Abordagens experimentais, especialmente com a criação de camundongos transgênicos, forneceram modelos valiosos para a análise da autotolerância, e muitos dos conceitos atuais são baseados nos estudos realizados com esses modelos Além disso, a identificação de genes que podem ser associados à autoimunidade em camundongos e humanos permitiu a dedução de alguns dos mecanismos críticos de autotolerância. Nas seções a seguir, discutiremos a tolerância central e periférica primeiro nas células T e a seguir nos linfócitos B, mas muitos aspectos dos processos são comuns a ambas as linhagens.
TOLERÂNCIA DOS LINFÓCITOS T
A tolerância dos linfócitos T CD4+ auxiliares (helper) é um mecanismo efetivo para prevenir a resposta imunológica contra os antígenos protéicos, uma vez que as células T auxiliares são indutores necessários tanto na resposta imunológica celular quanto na humoral a proteínas. Essa observação foi um grande estímulo para a realização de vários estudos sobre os mecanismos de tolerância das células T CD4+. Os imunologistas também desenvolveram modelos experimentais para o estudo da tolerância das células T CD4+ que forneceram um grande número de informações. Muitas das abordagens terapêuticas que estão sendo desenvolvidas para a indução de tolerância nos transplantes e antígenos próprios são alvos dessas células. Portanto, grande parte da discussão a seguir, especialmente sobre tolerância periférica, concentra-se nas células T CD4+. Sabe-se menos sobre tolerância periférica em células T CD8+, e isto é resumido ao final da seção.
Tolerância Central das Células T
Durante seu amadurecimento no timo, muitas células T imaturas que reconhecem antígenos com alta avidez são eliminadas e algumas das células sobreviventes na linhagem CD4+ se desenvolvem como células T reguladoras (Fig. 14-2). O processo de deleção, ou seleção negativa, de linfócitos T foi descrito no Capítulo 8, quando a maturação de células T no timo foi discutida. Esse processo afeta tanto células T restritas ao MHC de classe I e de classe II e, sendo assim, tem grande importância para a tolerância nas populações de linfócitos T CD8+ e CD4+. A seleção negativa dos timócitos é responsável pelo fato de o repertório das células T maduras, que deixam o timo e vão povoar os órgãos linfoides periféricos, não responder aos antígenos próprios que estão presentes no timo. Os dois principais fatores que determinam se um antígeno próprio específico irá induzir a seleção negativa nos timócitos autorreativos são a presença desse antígeno no timo, por expressão local ou por estar presente no sangue, e a afinidade dos receptores das células T (TCR) dos timócitos que reconhecem o antígeno. Assim as questões importantes que são relevantes para a seleção negativa são: quais antígenos próprios estão presentes no timo e como são mortas as células T imaturas que reconhecem estes antígenos.
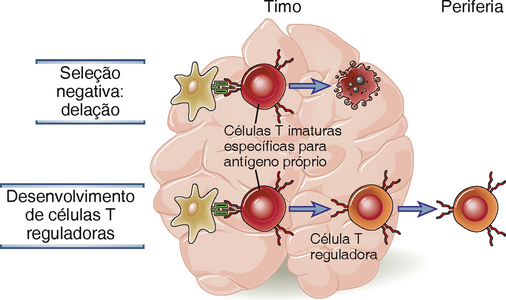
FIGURA 14-2 Tolerância central em célula T. Reconhecimento de antígenos próprios por células T imaturas no timo pode levar à morte das células (seleção negativa, ou deleção) ou ao desenvolvimento de células T reguladoras que entram em tecidos periféricos.
As proteínas próprias são processadas e apresentadas em associação às moléculas do MHC no timo pelas células apresentadoras de antígenos (APC). Os antígenos próprios apresentados no timo incluem diversas proteínas circulantes e associadas a células que são amplamente distribuídas nos tecidos. O timo tem também um mecanismo incomum para a expressão de antígenos proteicos que estão normalmente presentes em apenas alguns tecidos periféricos, de modo que células T imaturas que são específicas para estes antígenos podem ser eliminadas do repertório de células T em desenvolvimento. Alguns destes antígenos de tecidos periféricos são expressos em células epiteliais medulares do timo sob o controle da proteína reguladora autoimune (AIRE). Mutações no gene AIRE são a causa de uma doença autoimune em vários órgãos, chamada de síndrome poliendócrina autoimune (APS). Este grupo de doenças é caracterizado por lesão mediada por anticorpos e linfócitos em múltiplos órgãos endócrinos, incluindo as paratireóides, suprarrenais e ilhotas pancreáticas. Foi desenvolvido um modelo de APS em camundongos pela eliminação do gene AIRE, que recapitula muitas das características da doença humana. Estudos com camundongos mostraram que diversas proteínas que são produzidas em órgãos periféricos (como a insulina pancreática) também são normalmente expressas em baixos níveis em células epiteliais medulares do timo, e células T imaturas que reconhecem estes antígenos são eliminadas no timo. Na ausência de AIRE funcional (como nos pacientes e camundongos knockout), esses antígenos não são exibidos no timo, e as células T específicas para os antígenos escapam da deleção, maturam e entram na periferia, onde elas atacam os tecidos-alvo nos quais os antígenos são expressos independentemente do gene AIRE. A proteína AIRE pode funcionar como um fator de transcrição para promover a expressão de antígenos teciduais selecionados no timo. Ela é um componente de um complexo multiproteico que está envolvido no alongamento transcricional e no desenrolamento e remodelamento da cromatina. A AIRE também contribui para o processamento do pré-mRNA e induz o acúmulo de mRNA entrelaçados processados (ao contrário de mRNA não entrelaçados) de genes que codificam antígenos de tecidos periféricos. Também há evidências de mecanismos de deleção no timo que são independentes de AIRE.
Muitos timócitos imaturos com receptores de alta afinidade para antígenos próprios que encontram estes antígenos no timo morrem por apoptose. A seleção negativa ocorre nas células T duplamente positivas no córtex tímico ou em células unicamente positivas recém-geradas na medula. Nestes locais, timócitos imaturos com receptores de alta afinidade para antígenos próprios encontram estes antígenos e morrem por apoptose. A sinalização do receptor de célula T (TCR) em células T imaturas leva à ativação de uma proteína chamada Bim, que dispara a via mitocondrial de apoptose. Os mecanismos de apoptose são descritos mais adiante no capítulo, quando discutimos a deleção como um mecanismo de tolerância de célula T periférica. Obviamente, linfócitos imaturos e maduros interpretam sinais de receptores de antígenos de maneira diferente — os primeiros morrem e os últimos são ativados. A base bioquímica desta diferença não é conhecida.
Algumas células T CD4+ autorreativas que veem antígenos próprios no timo não são eliminadas, mas se diferenciam em células T reguladoras específicas para estes antígenos (Fig. 14-2). As células reguladoras saem do timo e inibem respostas contra tecidos próprios na periferia. Curiosamente, a deficiência da proteína AIRE, que interfere com a deleção das células T reativas com alguns antígenos no timo, não parece evitar o desenvolvimento de células T reguladoras de origem tímica específicas para os mesmos antígenos próprios. Esta observação sugere que os requisitos para deleção de células T e desenvolvimento de células T reguladoras no timo são diferentes, mas não se sabe o que determina a escolha entre morte celular e desenvolvimento de células T reguladoras. As características e funções de células T reguladoras são descritas mais adiante no contexto da tolerância periférica porque estas células suprimem respostas imunológicas na periferia.
Embora a importância da tolerância de células T central tenha sido claramente estabelecida em modelos animais, e a síndrome poliendócrina autoimune sugira que ela desempenha um papel fundamental na tolerância a alguns antígenos de tecidos periféricos, ainda não se sabe se uma falha na tolerância central contribui para doenças humanas autoimunes comuns.
Tolerância Periférica das Células T
A tolerância periférica é o mecanismo pelo qual as células T maduras que reconhecem antígenos próprios dos tecidos periféricos se tornam incapazes de responder subsequentemente a esses antígenos. Os mecanismos de tolerância periférica podem ser responsáveis pela tolerância das células T aos antígenos próprios dos tecidos específicos, especialmente aqueles que não são abundantes no timo. Os mesmos mecanismos podem induzir a não responsividade a formas tolerogênicos aos antígenos estranhos. Os mecanismos de tolerância periférica são anergia (não responsividade funcional), supressão, e deleção (morte da célula) (Fig. 14-3). Não sabemos se a tolerância a diferentes antígenos próprios é mantida por um ou outro mecanismo ou se todos estes mecanismos funcionam em cooperação para prevenir uma autoimunidade perigosa.
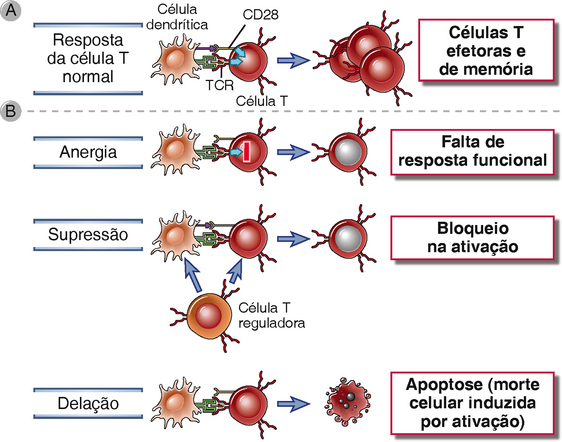
FIGURA 14-3 Mecanismos de tolerância periférica de célula T. Os sinais envolvidos em uma resposta imunológica normal (A) e os três principais mecanismos de tolerância periférica de célula T (B) são ilustrados.
Anergia (Não Responsividade Funcional)
Exposição de células T CD4+ maduras a um antígeno na ausência de coestimulação ou imunidade natural pode tornar as células incapazes de responder a esse antígeno. Neste processo, as células autorreativas não morrem, mas tornam-se não responsivas ao antígeno. Introduzimos anteriormente o conceito de que a ativação completa das células T requer o reconhecimento do antígeno pelo TCR (que fornece o sinal 1) e o reconhecimento dos coestimuladores, principalmente as moléculas B7-1 e B7-2, pelo CD28 (sinal 2) (Cap. 9). Sinal 1 (i. e., reconhecimento do antígeno) prolongado isoladamente pode levar à anergia. É provável que os antígenos próprios sejam exibidos a células T específicas na ausência de imunidade natural e coestimulação forte. A anergia induzida por antígenos foi demonstrada em uma variedade de modelos experimentais, incluindo estudos com clones de célula T expostos a antígenos in vitro (que foram a base para a definição original de anergia), experimentos nos quais os antígenos são administrados a camundongos sem adjuvantes, e estudos com camundongos transgênicos nos quais antígenos proteicos específicos são expressos durante toda a vida e reconhecidos por células T na ausência de inflamação e respostas imunológicas naturais que normalmente acompanham a exposição a micro-organismos. Em várias destas situações, as células T que reconhecem os antígenos tornam-se funcionalmente não responsivas e sobrevivem por dias ou semanas em um estado dormente.
A anergia resulta de alterações bioquímicas que reduzem a habilidade dos linfócitos de responder a sinais de seus receptores de antígeno (Fig. 14-4). Acredita-se que várias vias bioquímicas cooperam para manter este estado não responsivo.
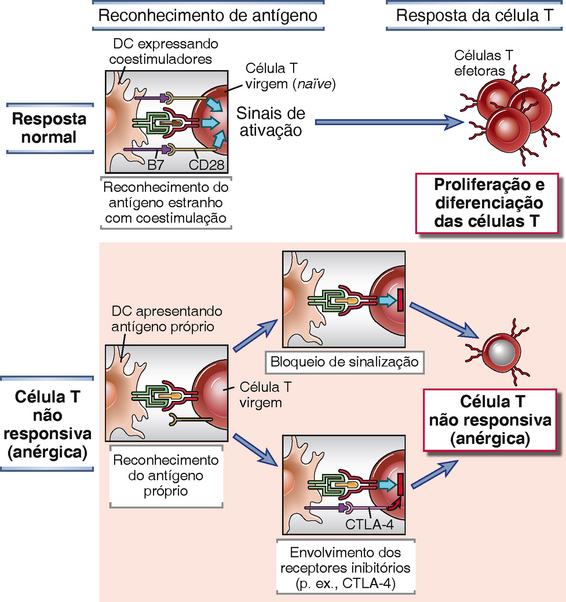
FIGURA 14-4 Mecanismos de anergia de célula T. As respostas de células T são induzidas quando as células reconhecem um antígeno apresentado por uma célula apresentadora de antígeno profissional (APC) e receptores ativadores nas células T (como CD28) reconhecem coestimuladores nas APCs (como B7). Se a célula T reconhece um antígeno próprio sem coestimulação, a célula T torna-se irresponsiva ao antígeno por causa de um bloqueio na sinalização do complexo TCR ou do envolvimento de receptores inibitórios (como CTLA-4). O bloqueio na sinalização pode ser o resultado do recrutamento de fosfatases para o complexo TCR ou a ativação de ligases de ubiquitina que degradam as proteínas sinalizadoras. A célula T permanece viável, mas é incapaz de responder ao antígeno próprio. DC, célula dendrítica.
• As células anérgicas mostram um bloqueio na transdução do sinal induzido pelo TCR. Os mecanismos deste bloqueio da sinalização não são totalmente compreendidos. Em diferentes modelos experimentais, ele é atribuível à expressão diminuída de TCR (talvez em virtude de degradação aumentada; ver a seguir) e ao recrutamento diminuído para o complexo TCR de moléculas inibidoras como tirosinas fosfatases.
• O reconhecimento de antígeno próprio pode ativar ubiquitinas ligases celulares, as quais podem ubiquitinar proteínas TCR associadas e dirigi-las para degradação proteolítica em proteassomas ou lisossomos. O resultado líquido é a perda destas moléculas de sinalização e ativação defeituosa das células T. Uma ligase de ubiquitina que é importante em células T é chamada de Cbl-b. Camundongos nos quais a Cbl-b foi colocada a knockout mostram proliferação espontânea de células T e manifestações de autoimunidade, sugerindo que esta enzima está envolvida na manutenção da não responsividade das células T aos antígenos próprios. Não se sabe por que o reconhecimento de antígenos próprios, que normalmente ocorre sem coestimulação forte, ativa estas ligases de ubiquitina, enquanto antígenos estranhos que são reconhecidos com coestimulação fazem muito menos ou nada.
• Quando células T reconhecem antígenos próprios, podem engajar receptores inibitórios da família CD28, cuja função é a de terminar respostas de célula T. No Capítulo 9, introduzimos o conceito geral que o resultado do reconhecimento de antígenos por células T, particularmente de células CD4+, é determinado por um equilíbrio entre o engajamento de receptores ativadores e inibitórios. Embora muitos receptores inibitórios tenham sido descritos, os dois cuja função fisiológica na autotolerância foi mais bem estabelecida são CTLA-4 e PD-1 (Fig. 9-5, Cap. 9).
CTLA-4, como o receptor ativador CD28, liga-se a moléculas B7. A CTLA-4 tem maior afinidade para moléculas B7 do que CD28, prevenindo assim coestimuladores nas APC de se engajar ao CD28; ele também pode remover moléculas B7 da superfície das APC (Fig. 14-5). Além disso, a CTLA-4 fornece sinais inibitórios que anulam os sinais disparados pelo TCR. Na verdade, a cauda citoplasmática da CTLA-4 tem um motivo potencialmente inibitório que pode contrapor-se aos sinais dependentes de ITAM do TCR e CD28. Como veremos mais adiante, a CTLA-4 também é um mediador da função inibitória de células T reguladoras. A importância da CTLA-4 na indução da tolerância é ilustrada pela verificação de que o camundongo knockout com supressão de CTLA-4 manifesta ativação descontrolada de linfócitos, com aumento maciço de linfonodos e do baço, e infiltrados linfocitários fatais em vários órgãos, sugerindo uma autoimunidade sistêmica. Em outras palavras, a eliminação deste único mecanismo de controle resulta em uma doença grave mediada por célula T, provavelmente por causa de defeitos tanto na anergia da célula T quanto na supressão por células T reguladoras. O bloqueio da CTLA-4 por anticorpos também desencadeia doenças autoimunes em animais, como a encefalomielite induzida pela imunização com antígenos de mielina e diabetes induzidos pela injeção de células T reativas com antígenos nas células β das ilhotas pancreáticas. Em testes clínicos do anticorpo anti-CTLA-4 para aumentar as respostas imunológicas a cânceres, alguns dos pacientes tratados desenvolvem manifestações de autoimunidade com inflamação em vários órgãos. Polimorfismos no gene CTLA4 são associados a várias doenças autoimunes em humanos, incluindo o diabetes tipo 1 e a doença de Graves. Todos esses achados indicam que a CTLA-4 funciona continuamente para manter as células T em ordem.
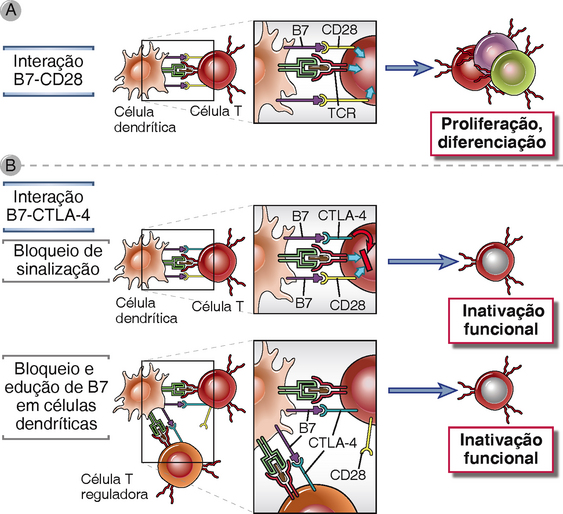
FIGURA 14-5 Mecanismos de ação da CTLA-4. A, O painel superior mostra a ativação de células T por reconhecimento de antígeno e coestimulação através de CD28. B, O painel inferior mostra os dois mecanismos de ação postulados para CTLA-4: entrega de sinais inibitórios que bloqueiam sinais mediados por TCR e CD28, e envolvimento de moléculas B7 nas APC de modo que estas ficam inacessíveis ao CD28. Note que as células T reguladoras (descritas mais neste capítulo) também podem usar CTLA-4 para bloquear B7 e assim inibir respostas imunológicas. Há evidências de que além de bloquear B7, a CTLA-4 pode remover estas moléculas da superfície da APC e internalizá-las (não mostrado).
Outro receptor inibitório da família CD28 é PD-1 (morte celular programada 1, assim chamada porque foi originalmente considerado envolvido na morte celular programada, mas que agora se sabe não ter um papel na apoptose de células T). O PD-1 reconhece dois ligantes, chamados PD-L1 e PD-L2; o PD-L1 é expresso nas APC e em muitas outras células de tecido, e o PD-L2 é expresso principalmente nas APC. O engajamento do PD-1 por qualquer um dos ligantes leva à inativação das células T. Camundongos nos quais PD1 foi colocado a nocaute desenvolvem doenças autoimunes, incluindo nefropatia semelhante a lúpus e artrite em diferentes raças puras. Os distúrbios autoimunes em camundongos knockout de PD-1 são menos graves do que naqueles com nocaute de CTLA-4. Foi postulado que as funções CTLA-4 controlam principalmente a ativação inicial da célula T em órgãos linfoides enquanto o PD-1 é mais importante para a limitação de respostas de células efetoras diferenciadas em tecidos periféricos.
Não se sabe como o equilíbrio entre a sinalização de receptores ativadores e inibitórios é regulada normalmente. Como mencionamos no Capítulo 9, uma explicação possível para o engajamento de CTLA-4 vs. CD28 por moléculas B7 é que as APC que apresentam antígenos próprios normalmente expressam baixos níveis de B7-1 e B7-2, o que é suficiente para engajar o receptor inibitório de alta afinidade CTLA-4. Em contrapartida, micro-organismos ativam as APC para aumentar a expressão de coestimuladores B7, e a CD28, que tem menor afinidade por moléculas B7 do que a CTLA-4, é engajada nestes níveis mais altos de expressão de B7. Isso pode explicar porque o reconhecimento de antígenos próprios pode pesar a balança na direção da CTLA-4, enquanto as infecções microbianas induzem relativamente mais sinais CD28.
Células dendríticas que são residentes nos órgãos linfoides e tecidos não linfoides podem apresentar antígenos próprios aos linfócitos T e manter a tolerância. Células dendríticas teciduais estão normalmente em um estado de repouso (imaturo) e expressam pouco ou nenhum coestimulador. Essas APC podem estar apresentando antígenos próprios constantemente sem ativar sinais, e as células T que reconhecem estes antígenos se tornam anérgicas. Também há evidências que células dendríticas em repouso tendem a promover o desenvolvimento de linfócitos T reguladores em vez de linfócitos efetores e de memória. Em contrapartida, células dendríticas que são ativadas por micro-organismos são as principais APC para a iniciação de respostas de células T (Cap. 6). Conforme discutiremos mais adiante, infecções e inflamações locais podem ativar células dendríticas residentes, levando à expressão aumentada de coestimuladores, destruição da tolerância e reações autoimunes contra os antígenos teciduais. As características das células dendríticas que as tornam tolerogênicas não foram definidas, mas provavelmente incluem a baixa expressão de coestimuladores. Há muito interesse em manipular as propriedades das células dendríticas de maneira a aumentar ou inibir respostas imunes para finalidades terapêuticas.
Supressão dos Linfócitos Autorreativos pelas Células T Reguladoras
O conceito de que alguns linfócitos poderiam controlar as respostas de outros linfócitos foi proposto há muito anos e logo seguido por demonstrações experimentais de populações de linfócitos T que suprimiam respostas imunológicas. Esses achados iniciais levaram a um enorme interesse sobre o tema, e “células T supressoras” tornaram-se um dos temas dominantes da imunologia nos anos 1970. Contudo, esse campo de pesquisa teve um histórico um tanto marcado, principalmente porque as tentativas iniciais de definir populações de células supressoras e seus mecanismos de ação foram bastante malsucedidas. Mais de 20 anos depois, a ideia teve um renascimento impressionante, com a aplicação de abordagens melhores para definir, purificar, e analisar populações de linfócitos T que inibem respostas imunológicas. Estas células são chamadas de linfócitos T reguladores; suas propriedades e funções são descritas a seguir.
Linfócitos T reguladores são um subconjunto de células T CD4+ cuja função é suprimir respostas imunológicas e manter a autotolerância (Fig. 14-6). A maioria destes linfócitos T reguladores CD4+ expressa altos níveis da cadeia α (CD25) do receptor de interleucina-2 (IL-2), mas não outros marcadores de ativação de célula T. Um fator de transcrição chamado FoxP3, um membro da família forkhead de fatores de transcrição, é crítico para o desenvolvimento e função da maioria das células T reguladoras. Camundongos com mutações no gene FOXP3 e camundongos nos quais este gene sofreu nocaute, desenvolvem uma doença autoimune multissistêmica associada a uma ausência de células T reguladoras CD25+. Uma rara doença autoimune em humanos chamada IPEX (síndrome de desregulação imune, poliendocrinopatia e enteropatia ligada ao X) é também associada à deficiência de células T reguladoras, e agora se sabe que é causada por mutações no gene FOXP3. Estes resultados estabelecem a importância das células T reguladoras para a manutenção da autotolerância. A recente onda de interesse em células T reguladoras é causada por uma maior compreensão de suas funções fisiológicas, bem como pela possibilidade de defeitos nestas células poderem resultar em diversas doenças autoimunes e, inversamente, as células T reguladoras poderem ser usadas para tratar doenças inflamatórias.
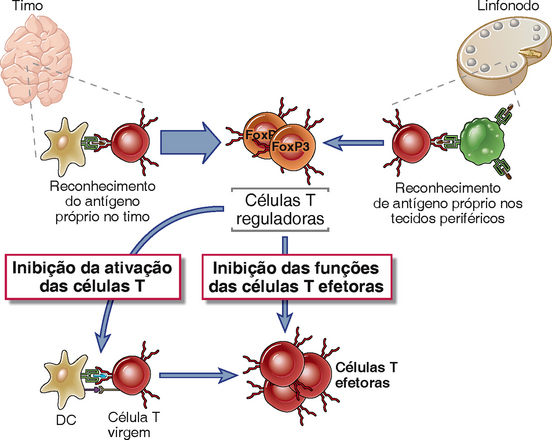
FIGURA 14-6 Células T reguladoras. Células T reguladoras são geradas pelo reconhecimento de antígenos próprios no timo (às vezes chamadas de células reguladoras naturais) e (provavelmente em menor grau) pelo reconhecimento de antígenos em órgãos linfoides periféricos (chamadas de células reguladoras induzíveis ou adaptativas). O desenvolvimento e sobrevivência destas células T reguladoras exige IL-2 e o fator de transcrição FoxP3. Em tecidos periféricos, células T reguladoras suprimem a ativação e as funções efetoras de outros linfócitos, autorreativos e potencialmente patogênicos.
Marcadores Fenotípicos e Heterogeneidade de Células T Reguladoras
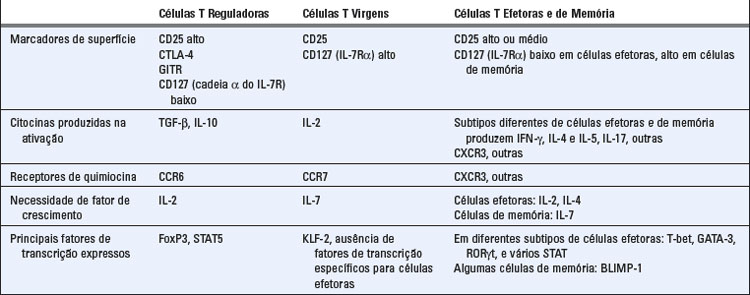
As células T reguladores são fenotipicamente diferentes de outras populações de linfócitos (Tabela 14-1). Apesar de numerosas populações de célula T terem sido descritas como possuidoras de atividade supressoras, o tipo de célula cuja função reguladora foi mais bem estabelecida foi a CD4+ FoxP3+ CD25alto. Ambas a FoxP3 e a CD25 são essenciais para a geração, manutenção e funcionamento destas células. Essas células geralmente expressam baixos níveis de receptores para IL-7 (CD127), e conforme previsto deste padrão de expressão de receptor, elas usam IL-2, mas não IL-7 como seu fator de crescimento e sobrevivência. Curiosamente, células T de memória têm a expressão do receptor e dependência do fator de crescimento opostos; elas normalmente são CD127alto e CD25baixo e dependem da IL-7 para sua manutenção. Células T reguladoras FoxP3+ geralmente também expressam altos níveis de CTLA-4, que é necessário para seu funcionamento (discutido mais adiante).
Geração e Manutenção de Células T Reguladoras
As células T reguladoras são geradas principalmente pelo reconhecimento de antígenos próprios no timo e pelo reconhecimento de antígenos próprios e estranhos nos órgãos linfoides periféricos. No timo, o desenvolvimento de células T reguladoras é um dos destinos das células T comprometidas com a linhagem CD4 que reconhece antígenos próprios; estas células derivadas do timo são às vezes chamadas de células T reguladoras naturais. Em órgãos linfoides periféricos, o reconhecimento de antígenos na ausência de respostas imunológicas naturais fortes favorece a geração de células reguladoras de linfócitos T CD4+ virgens (naïve), embora células T reguladoras também possam se desenvolver depois de reações inflamatórias. Essas células reguladoras geradas perifericamente já foram chamadas de adaptativas ou induzíveis porque elas podem ser induzidas a se desenvolver de células T CD4+ virgens como uma adaptação do sistema imunológico em resposta a certos tipos de exposição a antígenos. Previsivelmente, células reguladoras derivadas do timo são específicas para antígenos próprios porque estes são os antígenos encontrados principalmente no timo. Células reguladoras geradas perifericamente podem ser específicas para antígenos próprios ou estranhos. Também não está claro se tanto as células T reguladoras periféricas como as derivadas do timo contribuem para a manutenção da autotolerância ou se uma das populações é mais importante do que a outra para a prevenção da autoimunidade.
A geração e sobrevida das células T reguladoras são dependentes das citocinas TGF-β e IL-2. A cultura de células T virgens com anticorpos anti-TCR ativadores junto com TGF-β e IL-2 pode induzir o desenvolvimento de células reguladoras in vitro; estas às vezes também são chamadas de células T reguladoras induzidas. Em camundongos, a eliminação de TGF-β ou o bloqueio de sinais de TGF-β em células T leva a uma doença inflamatória sistêmica principalmente por causa de uma deficiência de células T reguladoras funcionais. TGF-β estimula a expressão de FoxP3, o fator de transcrição que leva à diferenciação de células T para a linhagem reguladora. Da mesma maneira, camundongos nos quais o gene para IL-2 ou para a cadeia α ou β do receptor de IL-2 sofreu nocaute desenvolvem autoimunidade, manifestada por doença intestinal inflamatória, anemia hemolítica autoimune, e múltiplos autoanticorpos (incluindo antieritrócitos e anti-DNA). Estes camundongos não têm um conjunto completo de células T reguladoras CD25+ FoxP3+, e sua doença pode ser corrigida pela recuperação destas células (fornecendo células de medula óssea de animais normais que podem gerar células FoxP3+). IL-2 promove a diferenciação de células T no subconjunto regulador e é também necessária para a sobrevida e manutenção desta população celular. IL-2 ativa o fator de transcrição STAT5, que pode aumentar a expressão de FoxP3 assim como de outros genes sabidamente envolvidos no funcionamento de células T reguladoras.
Populações específicas ou subconjuntos de células dendríticas podem ser especialmente importantes para estimular o desenvolvimento de células T reguladoras em tecidos periféricos. Existe alguma evidência que células dendríticas expostas ao ácido retinoico, o análogo da vitamina A, são indutoras de células T reguladoras, especialmente em tecidos linfoides associados à mucosa (Cap. 13).
Mecanismos de Ação de Células T Reguladoras
Células T reguladoras parecem suprimir respostas imunológicas em múltiplas etapas — na indução da ativação da célula T em órgãos linfoides e na etapa efetora destas respostas em tecidos. Apesar de diversos mecanismos de supressão terem sido descritos, os dois que são suportados pela maior quantidade de dados envolvem citocinas inibitórias e um efeito mediado por contato nas APC.
• Células T reguladoras produzem IL-10 e TGF-β ambos os quais inibem respostas imunológicas. A biologia destas citocinas é descrita em maiores detalhes a seguir.
• Células T reguladoras inibem a habilidade de estimulação de células T das APC. Um mecanismo proposto para esta ação é dependente da CTLA-4, que é expressa pelas células reguladoras FoxP3+ e parece ser necessária para seu funcionamento. Pode ser que a CTLA-4 em células reguladoras se ligue a moléculas B7 nas APC e bloqueie estas moléculas ou as remova através de sua internalização, resultando na disponibilidade reduzida de B7 e em uma inabilidade de fornecer coestimulação adequada para respostas imunológicas (Fig. 14-5).
Outros mecanismos de supressão por células T reguladoras que foram relatados incluem o consumo de IL-2, matando de fome os linfócitos que respondem a este fator de crescimento essencial, e a morte de T células responsivas.
Não foi estabelecido se todas as células reguladoras funcionam através de todos estes mecanismos ou se existem subpopulações que usam mecanismos diferentes para controlar respostas imunológicas. Na realidade, há evidências de que duas populações diferentes de células T reguladoras em humanos podem ser distinguidas pela expressão de FoxP3 ou pela produção de IL-10 (veja mais adiante), mas esta separação pode não ser absoluta.
Citocinas Inibitórias Produzidas por Células T Reguladoras
TGF-β e IL-10 estão envolvidas tanto na geração quanto no funcionamento de células T reguladoras. Estas citocinas são produzidas por e agem em muitos outros tipos de célula além das células reguladoras. Descrevemos aqui as propriedades e ações destas citocinas.
Fator de Transformação do Crescimento-β
TGF-β foi descoberto como um produto tumoral que promovia a sobrevivência de células tumorais in vitro. Na realidade, ele é uma família de moléculas muito próximas codificadas por genes distintos, comumente designados TGF-β1, TGF-β2 e TGF-β3. Células do sistema imunológico sintetizam principalmente o TGF-β1. O TGF-β1 é uma proteína homodimérica sintetizada e secretada por células T reguladoras CD4+, macrófagos ativados e muitos outros tipos de célula. Ele é sintetizado como um precursor inativo que é dividido proteoliticamente no complexo de Golgi e forma um homodímero. Esse homodímeiro de TGF-β1 maduro é secretado de forma latente em associação com outros polipeptídeos, que devem ser removidos extracelularmente pela digestão enzimática antes que a citocina possa se ligar aos receptores e exercer efeitos biológicos. O receptor de TGF-β1 consiste em duas proteínas diferentes, TGF-βRI e TGF-βRII, ambas as quais fosforilam fatores de transcrição chamados SMAD. Na ligação com a citocina, um domínio de cinase serina/treonina de TGF-βRI fosforila SMAD2 e SMAD3, que se transloca com SMAD4 para o núcleo, se liga aos promotores dos genes alvos e regula sua transcrição.
TGF-β tem diversas funções importantes e bastantes diferentes no sistema imunológico.
• TGF-β inibe a proliferação e as funções efetoras de células T e a ativação de macrófagos. TGF-β inibe a ativação clássica de macrófagos, mas é um dos mediadores secretados por macrófagos ativados alternativamente (Cap. 10). TGF-β também suprime a ativação de outras células, como neutrófilos e células endoteliais. Através destas ações inibitórias, a TGF-β funciona para controlar respostas imunológicas e inflamatórias. Camundongos nos quais o gene codificador de TGF-β1 sofre o nocaute ou nos quais a sinalização para codificação de TGF-β é bloqueada desenvolvem lesões inflamatórias descontroladas e linfoproliferação.
• TGF-β regula a diferenciação de subtipos funcionais diferentes de células T. Como descrito anteriormente, o desenvolvimento de células T reguladoras FoxP3+ periféricas depende de TGF-β. Entretanto, em combinação com citocinas produzidas durante respostas imunológicas naturais, como IL-1 e IL-6, o TGF-β promove o desenvolvimento do subconjunto TH17 de células T CD4+ em virtude de sua habilidade de induzir o fator de transcrição RORγt (Cap. 9). A habilidade do TGF-β de suprimir respostas imunológicas e inflamatórias, em parte pela geração de células T reguladoras, e também para promover o desenvolvimento de células TH17 inflamatórias na presença de outras citocinas é um exemplo interessante de como uma única citocina pode ter ações diversas e às vezes opostas, dependendo do contexto no qual ela é produzida. TGF-β também pode inibir o desenvolvimento de subconjuntos TH1 e TH2.
• TGF-β estimula a produção de anticorpos IgA induzindo células B a alternar para esse isotipo. IgA é o isotipo de anticorpo necessário para a imunidade das mucosas (Cap. 13).
• TGF-β promove o reparo do tecido depois que reações imunológicas e inflamatórias diminuem. Esta função é mediada principalmente pela habilidade da TGF-β de estimular a síntese de colágeno e a produção de enzimas modificadoras de matrizes por macrófagos e fibroblastos e pela promoção da angiogênese. Esta citocina pode desempenhar uma função patológica em doenças nas quais a fibrose é um componente importante, como a fibrose pulmonar e a esclerose sistêmica. Em reações fibróticas e de reparo, macrófagos ativados alternativamente podem ser uma das principais fontes de TGF-β.
Interleucina-10
A IL-10 é um inibidor de macrófagos ativados e de células dendríticas e está, portanto, envolvida no controle de reações da imunidade natural e da imunidade mediada por células. Ela é um membro de uma família de citocinas heterodiméricas, onde cada cadeia contém um domínio de feixe de seis hélices que se intercala com aquele da outra cadeia. Outros membros da família incluem IL-19, IL-20, IL-22, IL-24, IL-26 e IL-27. O receptor IL-10 pertence à família de receptores de citocinas do tipo II (semelhante ao receptor de interferons) e consiste em duas cadeias, que se associam a cinases JAK1 e TYK2 da família Janus e ativam STAT3. A IL-10 é produzida por muitas populações de células imunológicas, incluindo macrófagos ativados e células dendríticas, células T reguladoras e células TH1 e TH2. Como ela é produzida por e inibe as funções de macrófagos e de células dendríticas, ela é um exemplo excelente de regulador de feedback negativo. IL-10 também é produzida por alguns tipos de células não imunes (p. ex., queratinócitos).
Os efeitos biológicos da IL-10 resultam de sua habilidade de inibir muitas das funções de macrófagos ativados e células dendríticas.
• IL-10 inibe a produção de IL-12 por células dendríticas e macrófagos ativados. Como a IL-12 é um estímulo crítico para a secreção de IFN-γ, que tem função importante em reações imunológicas naturais e adaptativas mediadas por células contra micro-organismos intracelulares, a IL-10 funciona para suprimir todas essas reações. Na verdade, a IL-10 foi inicialmente identificada como uma proteína que inibia a produção de IFN-γ.
• A IL-10 inibe a expressão de coestimuladores e de moléculas do MHC de classe II em células dendríticas e macrófagos. Devido a estas ações, a IL-10 serve para inibir a ativação de células T e encerrar reações imunológicas mediadas por células.
Foi descrita uma rara doença autoimune herdada, na qual mutações no receptor IL-10 causam colite severa que se desenvolve logo nos primeiros meses de vida, antes de 1 ano de idade. Camundongos knockout com ausência de IL-10 também desenvolvem colite, provavelmente em consequência da ativação descontrolada de macrófagos reagindo a micro-organismos entéricos. Acredita-se que esta citocina é especialmente importante para o controle de reações inflamatórias em tecidos de mucosa, particularmente no trato gastrointestinal (Cap. 13).
O vírus Epstein-Barr contém um gene homólogo à IL-10 humana, e a IL-10 viral tem as mesmas atividades da citocina natural. Isso cria a possibilidade intrigante da aquisição do gene similar a IL-10 durante a evolução do vírus tenha lhe dado a habilidade de inibir a imunidade do hospedeiro, gerando assim uma vantagem de sobrevida no hospedeiro infectado.
Funções das Células T Reguladoras na Autotolerância e na Autoimunidade
A elucidação da base genética da doença IPEX e da doença similar em camundongos causada por mutações no gene FoxP3, descrita anteriormente, é prova convincente da importância das células T reguladoras na manutenção da autotolerância e da homeostase no sistema imunológico. Inúmeras tentativas estão sendo feitas para identificar defeitos no desenvolvimento ou função das células T reguladoras nas doenças autoimunes mais comuns em humanos, como doença intestinal inflamatória, diabetes tipo 1 e esclerose múltipla. Parece provável que defeitos na geração ou no funcionamento de células T reguladoras ou a resistência de células efetoras à supressão contribui significativamente para a patogênese de muitas doenças autoimunes. Também há enorme potencial para gerar células reguladoras e usá-las para controlar respostas imunes patológicas, e muitas tentativas estão em andamento para desenvolver tais terapias, particularmente para tratar a rejeição de transplantes (Cap. 16).
Deleção de Células T por Morte Celular Apoptótica
Os linfócitos T que reconhecem antígenos próprios sem inflamação ou que são repetidamente estimulados por antígenos morrem por apoptose. Há duas vias principais de apoptose em diversos tipos de célula (Fig. 14-7), ambas as quais foram implicadas na deleção de células T por antígenos próprios.
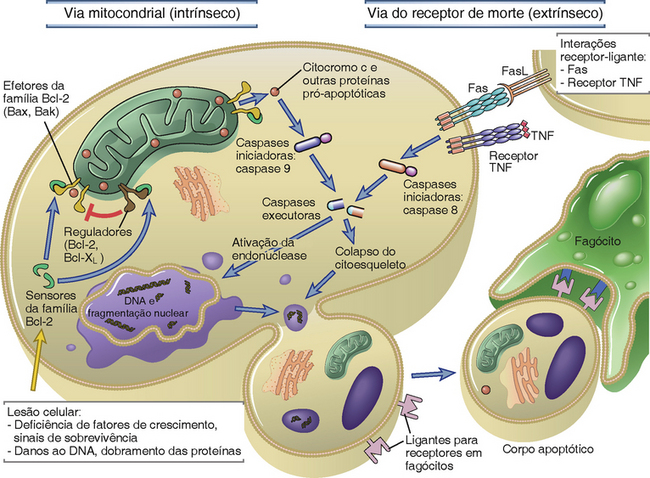
FIGURA 14-7 Vias de apoptose. A apoptose é induzida pelas vias mitocondrial e de receptor de morte, descritas no texto, que culminam na fragmentação da célula morta e na fagocitose dos corpos apoptóticos.
• A via mitocondrial (ou intrínseca) é regulada pela família de proteínas Bcl-2, cujo membro fundador, Bcl-2, foi descoberto como um oncogene em um linfoma de célula B e comprovadamente inibe a apoptose. Alguns membros desta família são proapoptóticos e outros são antiapoptóticos. A via é iniciada quando proteínas citoplasmáticas da família Bcl-2 que pertencem à subfamília “apenas BH3” (chamada assim porque contêm um domínio homólogo ao terceiro domínio conservado de Bcl-2) são induzidas ou ativadas em consequência de sinalização celular, privação do fator de crescimento, estímulos nocivos, ou danos ao DNA. Proteínas apenas-BH3 podem ser consideradas como “sensores” do estresse de células que pode se ligar a e influenciar a morte de efetores e reguladores. Em linfócitos, o mais importante destes sensores é uma proteína chamada Bim. A Bim ativada se liga a duas proteínas efetoras pró-apoptóticas da família Bcl-2 chamadas Bax e Bak, que se oligomerizam e se inserem na membrana mitocôndrica externa, levando a um aumento da permeabilidade mitocondrial. Os fatores de crescimento e outros sinais de sobrevivência induzem a expressão de membros antiapoptóticos da família Bcl-2, como Bcl-2 e Bcl-XL, que funcionam como reguladores de apoptose pela inibição de Bax e Bak, mantendo assim as mitocôndrias intactas. Proteínas apenas-BH3 também antagonizam Bcl-2 e Bcl-XL. Quando as células são privadas de sinais de sobrevivência, a mitocôndria começa a vazar devido às ações das proteínas BH3, Bax e Bak e à deficiência relativa de proteínas como Bcl-2 e Bcl-XL. O resultado é que muitos componentes mitocôndricos, incluindo o citocromo c, vazam para o citoplasma. Estas proteínas ativam enzimas citoplasmáticas chamadas caspases, inicialmente a caspase-9, que por sua vez cliva e ativa uma série de outras caspases que levam à fragmentação do DNA nuclear e a outras mudanças que culminam na morte apoptótica.
• Na via de receptores de morte (ou extrínseca) os receptores na superfície da célula homólogos aos receptores do fator de necrose tumoral (TNF) são engajados por seus ligantes, que são homólogos à citocina TNF. Os receptores se oligomerizam e ativam proteínas adaptadoras citoplasmáticas, que reúnem e clivam a caspase-8. A caspase-8 ativa cliva então uma série de outras caspases, resultando novamente em apoptose. Em muitos tipos de célula, a caspase-8 se cliva e ativa uma proteína apenas-BH3 que induz apoptose mitocôndrica. A via mitocondrial pode assim servir para amplificar a sinalização do receptor de morte.
Células que passam pela apoptose desenvolvem bolhas na membrana, e fragmentos do núcleo e do citoplasma se rompem em estruturas ligadas à membrana, chamadas corpos apoptóticos. Também ocorrem mudanças bioquímicas na membrana plasmática, incluindo a exposição de lipídios como a fosfatidilserina, que normalmente fica na face interna da membrana plasmática. Estas alterações são reconhecidas por receptores nos fagócitos, e células apoptóticas são rapidamente engolfadas e eliminadas, sem nunca ter elicitado uma resposta inflamatória do hospedeiro.
A melhor evidência para o envolvimento das duas vias apoptóticas na eliminação de linfócitos maduros autorreativos é que a ablação genética dos dois em camundongos resulta na autoimunidade sistêmica. Estas duas vias de morte podem funcionar de modos diferentes para manter a autotolerância. A morte celular que ocorre como consequência de exposição de células T maduras ao antígeno é às vezes chamada de morte celular induzida por ativação.
• As células T que reconhecem antígenos próprios sem coestimulação podem ativar a Bim, resultando em apoptose pela via mitocondrial. Em respostas imunológicas normais, os linfócitos respondentes recebem sinais do TCR, dos coestimuladores, e dos fatores de crescimento. Estes sinais estimulam a expressão de proteínas antipoptóticas da família Bcl-2(Bcl-2, Bcl-XL), prevenindo assim a apoptose e promovendo a sobrevivência da célula, o prelúdio necessário para a proliferação subsequente. Quando as células T reconhecem avidamente antígenos próprios, elas podem ativar a Bim diretamente, o que dispara a morte pela via mitocondrial, como descrito anteriormente. Ao mesmo tempo, devido à relativa falta de coestimulação e de fatores de crescimento, os membros antiapoptóticos da família Bcl-2, Bcl-2 e Bcl-XL, são expressos em baixos níveis e as ações de Bim, Bax, e Bak, portanto não são combatidas. A via mitocondrial de apoptose dependente de Bim também está envolvida na seleção negativa de células T autorreativas no timo (descrito anteriormente) e na fase de contração (declínio) de respostas imunológicas depois que o antígeno iniciador foi eliminado (Cap. 9).
• A estimulação repetida das células T resulta na coexpressão de receptores de morte e de seus ligantes, e o empenho dos receptores de morte desencadeia morte apoptótica. Nas células T CD4+, receptor de morte mais importante é Fas (CD95), e o seu ligante é o ligante de Fas (FasL). Fas é um membro da família de receptores TNF, e o FasL é homólogo ao TNF. Quando as células T são repetidamente ativadas, FasL é expresso na superfície celular, e se liga ao Fas de superfície na mesma célula ou em células T adjacentes. Isto ativa uma cascata de caspases, as quais finalmente causam a morte apoptótica das células. A mesma via de apoptose pode estar envolvida na eliminação de linfócitos B autorreativos (discutido adiante). Camundongos portadores de mutações homozigóticas dos genes codificando Fas ou ligante de Fas forneceram a primeira evidência concreta de que a falha da morte celular apoptótica resulta na autoimunidade. Estes camundongos desenvolvem uma doença autoimune sistêmica com autoanticorpos múltiplos e nefrite, parecida com o lúpus eritematoso sistêmico humano (Cap. 18). A linhagem de camundongo lpr (para linfoproliferação) produz baixos níveis de proteína Fas, e a linhagem gld (para doença linfoproliferativa generalizada) produz FasL com uma mutação pontual que interfere em sua função de sinalização. Acredita-se que a causa da autoimunidade seja o acúmulo de células autorreativas B e T auxiliares devido à falha na eliminação por apoptose na periferia. Crianças com uma doença fenotipicamente semelhante foram identificadas e provou-se que elas são portadoras de mutações no gene Fas ou em genes que codificam proteínas na via de morte mediada por Fas que resultam em uma falha na morte celular induzida por ativação. Essa doença é chamada de síndrome linfoproliferativa autoimune (ALPS).
Tolerância Periférica em Linfócitos T CD8+
Grande parte do nosso conhecimento sobre tolerância das células T periféricas é limitada às células T CD4+, muito pouco se sabe sobre os mecanismos de tolerância de células T CD8+ maduras. É possível que, se as células T CD8+ reconhecessem os peptídeos associados ao MHC de classe I sem coestimulação, imunidade natural, ou células T auxiliares, as células CD8+ se tornem anérgicas. Nessa situação, as células T CD8+ poderão encontrar o sinal 1 (antígeno) sem os segundos sinais, e o mecanismo da anergia poderá ser essencialmente o mesmo que para os linfócitos T CD4+. O papel do CTLA-4 e outros receptores inibitórios na indução de anergia nas células T CD8+ não está bem estabelecido. Células T reguladoras CD25+ podem inibir diretamente a ativação de células T CD8+ ou suprimir células auxiliares CD4+ que são necessárias para respostas CD8+ completas (Cap. 9). Células T CD8+ que são expostas a altas concentrações de antígenos próprios também podem passar por morte celular apoptótica.
Fatores que Determinam o Grau de Tolerância dos Antígenos Próprios
Estudos realizados em uma variedade de modelos experimentais mostraram que muitas características dos antígenos protéicos determinam se esses antígenos induzirão a ativação ou a tolerância da célula T (Tabela 14-2). Os antígenos próprios têm várias propriedades especiais que os tornam tolerogênicos. Alguns antígenos próprios estão presentes no timo, e estes antígenos podem induzir a seleção negativa ou desenvolver células T reguladoras. Na periferia, antígenos próprios, que geralmente são expressos por longos períodos ou pela vida inteira, são capazes de engajar receptores de antígeno por períodos prolongados e são normalmente expostos a linfócitos sem inflamação ou imunidade natural. Nessas condições, as APC expressam pouco ou nenhum coestimulador, e o reconhecimento do antígeno poderá não provocar resposta (ignorância) ou induzir anergia, morte celular, ou células T reguladoras. Um conceito geral que surgiu foi que a ativação do receptor da célula T, na ausência de imunidade natural e de inflamação, tende a disparar um ou mais dos mecanismos de tolerância periférica, enquanto a imunidade natural, a coestimulação e as citocinas pendem a balança na direção da proliferação de célula T e da diferenciação em células efetoras e de memória. Nossa compreensão dos mecanismos que conectam os sinais que uma célula T recebe no momento do reconhecimento do antígeno ao destino da célula T permanece incompleta. Esses conceitos são, em grande parte, baseados em modelos experimentais nos quais antígenos são administrados aos camundongos ou são produzidos como transgenes expressos nestes animais. Um dos desafios contínuos neste campo é definir os mecanismos pelos quais vários antígenos próprios normalmente expressos induzem a tolerância, especialmente em seres humanos.
TABELA 14-2 Fatores que Determinam a Imunogenicidade e Tolerogenicidade de Antígenos Proteicos
| Fator | Características que Favorecem a Estimulação de Respostas Imunológicas | Características que Favorecem a Tolerância |
|---|---|---|
| Persistência | Vida curta (eliminada por resposta imunológica) | Prolongado |
| Portal de entrada; localização | Subcutânea, intradérmica; ausente em órgãos primários | Intravenosa, mucosal; presente em órgãos primários |
| Presença de adjuvantes | Antígenos com adjuvantes: estimulam células T auxiliadoras | Antígenos sem adjuvantes: não imunogênico ou tolerogênico |
| Propriedade de células apresentadoras de antígenos | Altos níveis de coestimuladores | Baixos níveis de coestimuladores e citocinas |
TOLERÂNCIA DOS LINFÓCITOS B
A tolerância nos linfócitos B é necessária para manter a não responsividade aos antígenos próprios timo-independentes, como os polissacarídeos e os lipídeos. A tolerância da célula B também exerce um papel na prevenção das respostas de anticorpo aos antígenos proteicos. Estudos experimentais revelaram mecanismos múltiplos pelos quais o encontro com antígenos próprios pode abortar a maturação e a ativação de células B.
Tolerância Central das Células B
Os linfócitos B imaturos que reconhecem os antígenos próprios com alta afinidade na medula óssea são eliminados ou mudam a sua especificidade. Os mecanismos de tolerância central de células B foram descritos, principalmente, em modelos experimentais (Fig. 14-8).
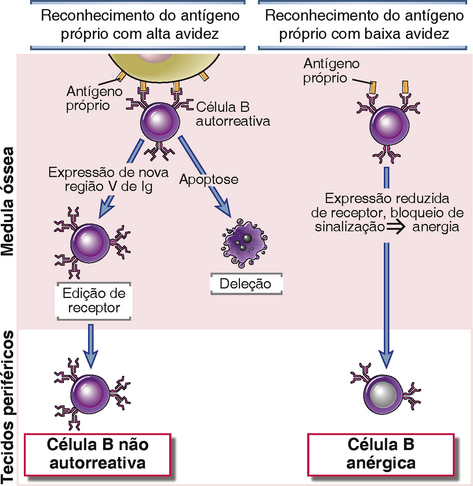
FIGURA 14-8 Tolerância central em células B. Células B imaturas que reconhecem antígenos próprios na medula óssea com alta avidez (p. ex., quantidades multivalentes de antígenos em células) morrem por apoptose ou alteram a especificidade de seus receptores de antígenos (edição de receptor). O fraco reconhecimento de antígenos próprios na medula óssea pode levar à anergia (desativação funcional) das células B.
• Edição de receptores. Se células B imaturas reconhecem antígenos próprios que estão presentes em alta concentração na medula óssea e especialmente se o antígeno for exposto de forma multivalente (p. ex., na superfície celular), muitos receptores de antígenos em cada célula B são cruzados, passando assim sinais fortes às células. Uma consequência desta sinalização é que as células B reativam seus genes RAG1 e RAG2 e iniciam uma nova rodada de recombinação VJ no lócus da cadeia leve κ da imunoglobulina (Ig). Um segmento Vκ da unidade VκJκ já rearranjada mais acima é unido a um Jκ mais abaixo na cadeia. Como consequência, o éxon VκJκ rearranjado anteriormente na célula B autorreativa imatura é deletado e uma nova cadeia leve de Ig é expressada, criando assim um receptor de célula B com uma nova especificidade. Este processo é conhecido como edição de receptor (Cap. 8) e é um importante mecanismo para eliminar a autorreatividade do repertório das células B maduras. Se o rearranjo da cadeia leve editada não for produtivo, o rearranjo pode prosseguir no lócus κ no outro cromossomo, e se este não for produtivo, pode haver rearranjos nos loci da cadeia leve λ em seguida. Uma célula B expressando uma cadeia leve λ frequentemente é uma célula que passou por edição de receptor.
• Deleção. Se a edição falhar, as células B imaturas podem ser eliminadas (i. e., elas morrem por apoptose). Os mecanismos de deleção não são bem definidos.
• Anergia. Se células B em desenvolvimento reconhecem antígenos próprios fracamente (p. ex., se o antígeno é solúvel e não faz ligações cruzadas com muito receptores de antígeno ou se os receptores da célula B reconhecem o antígeno com baixa afinidade), as células tornam-se funcionalmente não responsivas (anérgicas) e deixam a medula óssea neste estado irresponsivo. A anergia é causada pela infrarregulação da expressão do receptor de antígeno e também por um bloqueio na sinalização desse receptor.
Tolerância Periférica das Células B
Os linfócitos B maduros que reconhecem antígenos próprios nos tecidos periféricos na ausência de células T auxiliares específicas podem se tornar funcionalmente irresponsivos ou morrer por apoptose (Fig. 14-9). Sinais de células T auxiliares podem estar ausentes se estas células T forem eliminadas ou anérgicas ou se os antígenos próprios forem antígenos não proteicos. Como os antígenos próprios não obtêm respostas imunológicas naturais, as células B também não encontrarão nenhuma das citocinas ou outros sinais que são induzidos durante estas respostas. Portanto, como nas células T, o reconhecimento de antígenos sem estímulos adicionais resulta em tolerância. Mecanismos de tolerância periférica também eliminam clones autorreativos de célula B que podem ser gerados como uma consequência da mutação somática em centros germinais.
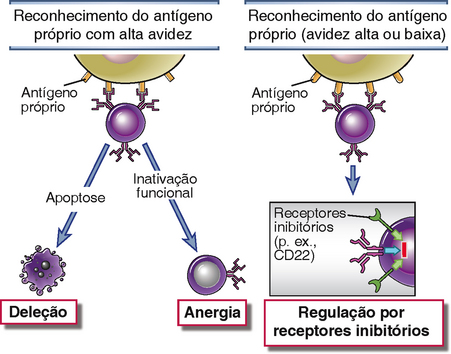
FIGURA 14-9 Tolerância periférica em células B. Células B que encontram antígenos próprios em tecidos periféricos tornam-se anérgicas ou morrem por apoptose. Em algumas situações, o reconhecimento de antígenos próprios pode disparar receptores inibitórios que evitam a ativação de células B.
• Anergia e deleção. Algumas células B autorreativas que são estimuladas repetidamente por antígenos próprios tornam-se irresponsivas a ativação posterior. Estas células exigem níveis mais elevados do fator de crescimento BAFF/BLys para sua sobrevida (Cap. 11) e não podem competir eficientemente com células B virgens normais dependentes de BAFF pela sobrevida em folículos linfoides. Em consequência, aquelas células B que encontraram antígenos próprios têm vida encurtada e são eliminadas mais rapidamente do que células que não reconheceram antígenos próprios. Células B que se ligam com alta avidez a antígenos próprios na periferia também podem passar por morte apoptótica pela via mitocondrial independentemente da dependência do fator de crescimento.
A taxa elevada de mutação somática de genes Ig que ocorre em centros germinais corre o risco de gerar células B autorreativas (Cap. 11). Estas células B podem ser ativamente eliminadas pela interação de FasL em células T auxiliares com Fas nas células B ativadas. A mesma interação foi descrita antes como um mecanismo para a morte de células T autorreativas. A falha desta via de tolerância periférica de célula B pode contribuir para a autoimunidade que é causada por mutações nos genes Fas e FasL em camundongos, e em pacientes com a síndrome linfoproliferativa autoimune mencionada anteriormente.
• Sinalização por receptores inibitórios. Células B que reconhecem antígenos próprios com baixa afinidade podem não conseguir responder por meio do envolvimento de vários receptores inibitórios. A função destes receptores inibitórios é de estabelecer um limiar para ativação da célula B, que permite respostas a antígenos estranhos com célula T auxiliar ou imunidade natural, mas não permite respostas a antígenos próprios. Esse mecanismo de tolerância periférica foi revelado em estudos mostrando que camundongos com defeitos na SHP-1 tirosina fosfatase ou no receptor inibitório CD22 desenvolvem autoimunidade. Motivos ITIM na cauda citoplasmática de CD22 são fosforilados por Lyn, e este receptor inibitório então recruta SHP-1, atenuando desta maneira a sinalização do receptor de célula B. Contudo, não se sabe quando receptores inibitórios como o CD22 são envolvidos e quais ligantes eles reconhecem.
Já se sabe muito sobre os mecanismos de tolerância em linfócitos T e B, em grande parte devido ao uso de modelos animais como camundongos geneticamente modificados. A aplicação desses conhecimentos na compreensão dos mecanismos da tolerância aos diferentes antígenos próprios em indivíduos normais e da definição do por que a tolerância falha, dando origem a doenças autoimunes, é uma área de intensa investigação.
TOLERÂNCIA INDUZIDA POR ANTÍGENOS PROTEICOS ESTRANHOS
Os antígenos estranhos podem ser administrados de modo que induzam preferencialmente tolerância em vez de respostas imunológicas. Entender como induzir a tolerância pela administração de antígenos é a chave para o desenvolvimento de tolerância a antígenos específicos como uma estratégia de tratamento para doenças imunológicas. Em geral, os antígenos proteicos administrados de maneira subcutânea ou intradérmica com adjuvantes favorecem a imunidade, enquanto altas doses do antígeno administradas de forma sistêmica, sem adjuvante, tendem a induzir tolerância. A razão provável para isso é que os adjuvantes estimulam as respostas imunológicas naturais e a expressão dos coestimuladores nas APC, e na ausência desses sinais secundários, as células T que reconhecem os antígenos podem tornar-se anérgicas ou morrer ou podem se diferenciar em células reguladoras. Muitas outras características dos antígenos, e as vias pelas quais eles são administrados, podem influenciar o equilíbrio entre imunidade e tolerância (Tabela 14-2).
A administração oral de um antígeno proteico leva frequentemente à supressão das respostas imunológicas sistêmicas humorais e mediadas por células com o mesmo antígeno. Este fenômeno, chamado tolerância oral, foi discutido no Capítulo 13.
Algumas infecções sistêmicas (p. ex., com vírus) podem iniciar uma resposta imunológica, mas a resposta é diminuída antes que o vírus seja removido, resultando em um estado de infecção persistente. Nesta situação, clones de célula T vírus-específicos estão presentes, mas não respondem normalmente e são incapazes de erradicar a infecção. Este fenômeno foi chamado de exaustão clonal, sugerindo que os clones de linfócitos antígeno-específicos dão uma resposta inicial, mas depois ficam anérgicos, ou “esgotados”. Há algumas evidências que a exaustão clonal é causada pela suprarregulação de receptores inibitórios como o PD-1 em células T CD8+ vírus-específicas. Este fenômeno foi visto em pacientes infectados com o vírus da imunodeficiência humana (HIV) e em modelos animais de infecção viral crônica. Não se sabe como alguns micro-organismos suprarregulam a expressão de moléculas inibitórias em células T. O esgotamento clonal pode favorecer a persistência viral e é, portanto, um mecanismo de evasão imunológica usado por alguns patógenos. A compreensão deste processo pode até abrir novos caminhos para intervenções terapêuticas em algumas doenças virais crônicas, como o tratamento com anticorpos bloqueadores de PD-1.
PATOGÊNESE DA AUTOIMUNIDADE
A possibilidade do sistema imunológico de um indivíduo poder reagir contra antígenos autólogos e causar dano ao tecido foi percebida por imunologistas a partir do momento em que a especificidade do sistema imunológico para antígenos estranhos foi reconhecida. No início dos anos 1900, Paul Ehrlich criou a frase melodramática “horror autotóxico” para reações imunológicas nocivas (“tóxicas”) contra o próprio. Autoimunidade é uma importante causa de doença em humanos e estima-se que afete de 2% a 5% da população dos EUA. O termo autoimunidade é usado com frequência erroneamente para qualquer doença onde reações imunológicas acompanham lesões ao tecido, mesmo que possa ser difícil ou impossível estabelecer uma função para respostas imunológicas contra antígenos próprios na causa destas desordens. Como a inflamação é um componente proeminente nestas doenças, elas às vezes são agrupadas como doenças inflamatórias imunologicamente mediadas, o que não implica que a resposta patológica é dirigida contra antígenos próprios (Cap. 18).
As questões fundamentais sobre a autoimunidade são como a autotolerância falha e como os linfócitos autorreativos são ativados. São necessárias respostas para estas questões para entender a etiologia e a patogênese de doenças autoimunes, que é um grande desafio na imunologia. Nossa compreensão da autoimunidade melhorou muito nas últimas duas décadas, principalmente devido ao desenvolvimento de modelos animais informativos destas doenças, a identificação de genes que podem predispor à autoimunidade, e a melhores métodos de análise de respostas imunológicas em humanos. Vários conceitos gerais importantes surgiram dos estudos sobre a autoimunidade.
• A autoimunidade resulta de uma falha dos mecanismos de autotolerância em células T ou B, o que pode levar a um desequilíbrio entre a ativação de linfócitos e os mecanismos de controle. O potencial para autoimunidade existem todos os indivíduos porque algumas das especificidades geradas aleatoriamente de clones de linfócitos em desenvolvimento podem ser para antígenos próprios, e muitos antígenos próprios são facilmente acessíveis aos linfócitos. Conforme discutido anteriormente, a tolerância a antígenos próprios é normalmente mantida por processos de seleção que previnem a maturação de alguns linfócitos específicos para antígenos próprios e por mecanismos que inativam ou deletam linfócitos autorreativos que maturam. A perda de autotolerância pode ocorrer se linfócitos autorreativos não forem eliminados ou inativados durante ou depois de sua maturação e se as APC são ativadas de modo que os antígenos próprios são apresentados ao sistema imunológico de uma maneira imunogênica. Alguns dos mecanismos gerais que são associados às reações autoimunes são os seguintes:
Em nossa discussão anterior dos mecanismos de autotolerância, fizemos referência a muitas destas anomalias para ilustrar como a autotolerância pode falhar, resultando na autoimunidade. Voltaremos a discutir estas aberrações imunológicas como a base da autoimunidade a seguir e no Capítulo 18, quando consideraremos doenças selecionadas.
Recentemente, muita atenção tem sido focada na função das células T na autoimunidade, por duas razões principais. Primeira, as células T auxiliares são os reguladores-chave de todas as respostas imunológicas a proteínas, e a maioria dos antígenos próprios implicados em doenças autoimunes são proteínas. Segunda, diversas doenças autoimunes são geneticamente ligadas ao MHC (o complexo HLA em humanos), e a função das moléculas do MHC é apresentar antígenos peptídicos a células T. Uma falha na autotolerância em linfócitos T pode resultar em doenças autoimunes nas quais os danos ao tecido são causados por reações imunológicas mediadas por células. Anomalias de células T auxiliares também podem levar à produção de autoanticorpos, pois as células T auxiliares são necessárias para a produção de anticorpos de alta afinidade contra antígenos proteicos.
• Os principais fatores que contribuem para o desenvolvimento da autoimunidade são a suscetibilidade genética e as causas ambientais, como infecções e lesões teciduais locais. Genes de suscetibilidade podem interromper os mecanismos de autotolerância, e infecção ou necrose nos tecidos promove a entrada de linfócitos autorreativos e a ativação destas células, resultando em lesões teciduais (Fig. 14-10). Infecções e lesões teciduais podem também alterar a forma em que os antígenos próprios são expostos ao sistema imunológico, levando à falha da autotolerância e à ativação de linfócitos autorreativos. As funções destes fatores no desenvolvimento da autoimunidade são discutidas mais adiante.
• Doenças autoimunes podem ser sistêmicas ou órgão-específicas, dependendo da distribuição dos antígenos próprios que são reconhecidos. Por exemplo, a formação de complexos imunológicos circulantes, compostos de nucleoproteínas próprias e anticorpos específicos normalmente produza doenças sistêmicas, como o lúpus eritematoso sistêmico (SLE). Em contrapartida, as respostas de autoanticorpos ou de células T contra antígenos próprios com distribuição tecidual restrita levam a doenças órgão-específicas, como miastenia grave, diabetes tipo 1 e esclerose múltipla.
• Vários mecanismos efetores são responsáveis por lesões teciduais em doenças autoimunes diferentes. Estes mecanismos incluem complexos imunológicos, autoanticorpos circulantes, e linfócitos T autorreativos e são discutidos no Capítulo 18. As características clínicas e patológicas da doença são geralmente determinadas pela natureza da resposta autoimune dominante.
• Doenças autoimunes tendem a ser crônicas, progressivas, e autoperpetuadora. As razões para estas características são que os antígenos próprios que disparam essas reações são persistentes, e uma vez iniciada uma resposta imunológica, muitos mecanismos de amplificação que perpetuam a resposta são ativados (Fig. 14-11). Além disso, uma resposta iniciada contra um antígeno próprio que lesiona tecidos pode resultar na liberação e alterações de outros antígenos teciduais, na ativação de linfócitos específicos para estes outros antígenos, e na exacerbação da doença. Este fenômeno é chamado de expansão dos epítopos, e pode explicar por que uma vez que uma doença autoimune se desenvolveu, ela pode tornar-se prolongada e autoperpetuadora.
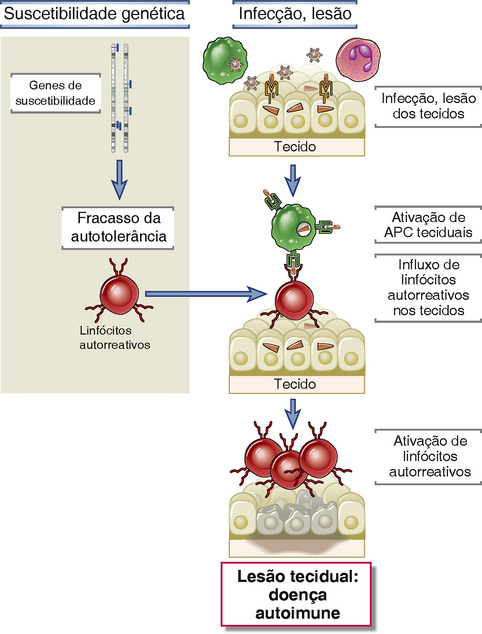
FIGURA 14-10 Mecanismos postulados de autoimunidade. Neste modelo proposto de uma doença autoimune específica a um órgão, mediada por célula T, vários loci genéticos podem conceder suscetibilidade à autoimunidade, em parte influenciando a manutenção da autotolerância. Fatores ambientais, como infecções e outros estímulos inflamatórios, promovem a entrada dos linfócitos nos tecidos e a ativação de células T autorreativas, resultando em lesões teciduais.
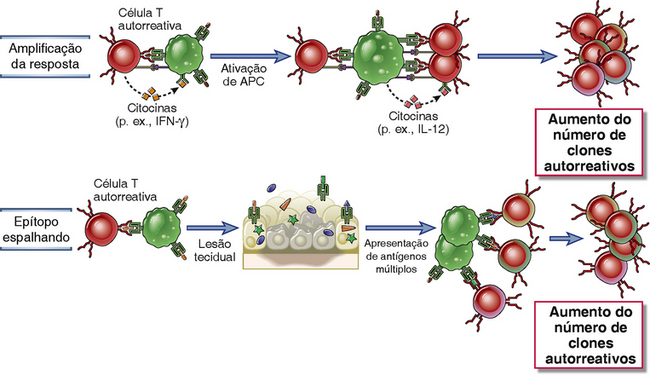
FIGURA 14-11 Mecanismos de cronicidade de doenças autoimunes. Depois do desenvolvimento de uma reação autoimune, mecanismos de amplificação (como citocinas, mostradas como um exemplo ilustrativo) promovem a ativação de linfócitos autorreativos, e a liberação de antígenos próprios de células e tecidos danificados leva à expansão dos epítopos.
Na seção a seguir, descrevemos os princípios gerais da patogênese de doenças autoimunes, com ênfase nos genes da suscetibilidade, infecções e outros fatores que contribuem para o desenvolvimento da autoimunidade. A patogênese e as características de algumas doenças autoimunes ilustrativas são descritas no Capítulo 18.
Base Genética da Autoimunidade
Dos estudos iniciais de doenças autoimunes em pacientes e em animais experimentais, verificou-se que estas doenças têm forte componente genético. Por exemplo, o diabetes tipo 1 tem concordância de 35% a 50% em gêmeos monozigóticos e de 5% a 6% em gêmeos dizigóticos, e outras doenças autoimunes, mostram evidências semelhantes de contribuição genética. Análises de ligações em famílias, estudos de associação genômica abrangentes e esforços de ressequenciamento em grande escala têm revelado novas informações sobre os genes que podem desempenhar funções causais no desenvolvimento da autoimunidade e de doenças inflamatórias crônicas. Com base nestes estudos, diversas características gerais da suscetibilidade genética ficaram aparentes.
As doenças autoimunes, em sua maioria, são traços poligênicos complexos, onde os indivíduos afetados herdam polimorfismos genéticos múltiplos que contribuem para a suscetibilidade à doença, e estes genes agem com fatores ambientais para causar as doenças. Alguns destes polimorfismos são associados a várias doenças autoimunes, sugerindo que os genes causadores influenciam os mecanismos gerais da regulação imunológica e da autotolerância. Outros loci são associados a doenças específicas, sugerindo que podem afetar os danos a órgãos ou linfócitos autorreativos de especificidades particulares. Cada polimorfismo genético faz uma pequena contribuição para o desenvolvimento de doenças autoimunes específicas e também é encontrado em indivíduos saudáveis, mas com menor frequência do que em pacientes com as doenças. Postula-se que em pacientes individuais, tais polimorfismos múltiplos são co-herdados e são conjuntamente responsáveis pelo desenvolvimento da doença. Compreender a interação dos genes múltiplos um com o outro e com os fatores ambientais é um dos desafios constantes deste campo.
Os genes mais bem caracterizados associados a doenças autoimunes e nosso conhecimento atual de como eles podem contribuir para a perda da autotolerância são descritos aqui.
Associação de Alelos do MHC e Autoimunidade
Entre os genes que são associados à autoimunidade, as associações mais fortes são com genes do MHC. Na realidade, em muitas doenças autoimunes, como o diabetes tipo 1, foram identificados 20 ou 30 genes associados à doença; na maior parte destas doenças, o lócus HLA contribui sozinho para metade ou mais da suscetibilidade genética. Tipagem HLA de grandes grupos de pacientes com várias doenças autoimunes mostrou que alguns alelos HLA ocorrem com maior frequência nestes pacientes do que na população geral. Destes estudos, podemos calcular a razão de probabilidade para o desenvolvimento de uma doença em indivíduos que herdam vários alelos HLA (frequentemente chamado de risco relativo na literatura mais antiga) (Tabela 14-3). A associação mais forte deste tipo é entre a espondilite anquilosante, uma doença inflamatória, presumivelmente autoimune, das juntas vertebrais, e o alelo HLA classe I B27. Indivíduos que são positivos para HLA-B27 têm uma razão de probabilidade de mais 100 para o desenvolvimento de espondilite anquilosante. Nem o mecanismo desta doença nem a base de sua associação com o HLA-B27 são conhecidos. A associação de alelos classe II HLA-DR e HLA-DQ a doenças autoimunes tem recebido muita atenção, principalmente porque as moléculas do MHC de classe II estão envolvidas na seleção e ativação de células T CD4+, e células T CD4+ regulam as respostas imunológicas humorais e celulares a antígenos proteicos.
TABELA 14-3 Associação de Alelos HLA e Doença Autoimune
| Doença | Alelo HLA | Razão de Probabilidade1 |
|---|---|---|
| Artrite reumatoide (Anticorpo anti-CCP positivo)2 | Alelo DRB1, 1 SE3 | 4 |
| Alelos DRB1, 2 SE | 12 | |
| Diabetes tipo 1 | Haplótipo DRB1*0301- DQA1*0501-DQB1*0201 | 4 |
| Haplótipo DRB1*0401- DQA1*0301-DQB1*0302 | 8 | |
| Heterozigotos DRB1*0301/0401 | 35 | |
| Esclerose múltipla | DRB1*1501 | 3 |
| Lúpus eritematoso sistêmico | DRB1*0301 | 2 |
| DRB1*1501 | 1,3 | |
| Espondilite anquilosante | B*27 (principalmente B*2705 e *2702) | 100-200 |
| Doença celíaca | Haplótipo DQA1*0501- DQB1*0201 | 7 |
1 A razão de probabilidade aproxima valores de risco aumentado da doença associada à herança de alelos HLA específicos. Os dados são de populações de ascendência europeia.
2 Anti-CCP, anticorpos dirigidos contra peptídeos citrulinados cíclicos. Dados são de pacientes que testam positivo para estes anticorpos no soro.
3 SE se refere ao epítopo compartilhado, assim chamado porque é uma sequência de consenso na proteína DRB1 (posições 70-74, presente em vários alelos DRB1). (Cortesia de Dr. Michelle Fernando, London.)
Diversas características da associação de alelos HLA e doenças autoimunes são relevantes.
• Uma associação HLA-doença pode ser identificada por tipagem sorológica de um lócus HLA, mas a associação real pode ser com outros alelos que estão ligados ao alelo tipado e herdados juntos. Por exemplo, indivíduos com um alelo HLA-DR específico (hipoteticamente DR1) podem ter uma maior probabilidade de herdar um alelo HLA-DQ específico (hipoteticamente DQ2) do que a probabilidade de herdar estes alelos separadamente e aleatoriamente (ou seja, em equilíbrio) na população. Tal herança é um exemplo de desequilíbrio de ligação. Pode-se descobrir que uma doença é associada a DR1 por tipagem HLA, mas a associação causal pode realmente ser com o DQ2 co-herdado. Esta realização enfatizou o conceito de “haplótipos estendidos do HLA”, que se refere a conjuntos de genes ligados, tanto genes HLA clássicos como genes não HLA adjacentes, que tendem a ser herdados juntos como uma única unidade.
• Em muitas doenças autoimunes, os polimorfismos de nucleotídeos associados à doença codificam aminoácidos nas fendas de ligação de peptídeos em moléculas do MHC. Esta descoberta não é surpreendente porque resíduos polimorfos de moléculas do MHC são localizados dentro das e adjacentes às fendas, e a estrutura das fendas é a chave determinante de ambas as funções das moléculas MHC; a saber, apresentação de antígeno e reconhecimento por células T (Cap. 6). Estes resultados suportam o conceito geral que moléculas do MHC influenciam o desenvolvimento da autoimunidade pelo controle da seleção e ativação de células T.
• Sequências HLA associadas a doenças são encontradas em indivíduos saudáveis. De fato, se todos os indivíduos portadores de um alelo HLA específico associado à doença são monitorados prospectivamente, a maioria jamais terá a doença. Portanto, a expressão de um gene HLA específico não é por si só a causa de uma doença autoimune, mas ele pode ser um dos vários fatores que contribuem para a autoimunidade.
Os mecanismos subjacentes à associação de alelos do HLA específicos e várias doenças autoimunes ainda não estão claros. Quando são notadas associações positivas de alelos do MHC e a doença, a molécula do MHC associada à doença pode apresentar um peptídeo próprio e ativar células T patogênicas, e isso foi estabelecido em alguns casos. Quando se demonstra que um alelo específico é protetor (associação negativa com a doença), supõe-se que este alelo pode induzir a seleção negativa de algumas células T em desenvolvimento e potencialmente patogênicas, criando assim um “buraco no repertório”, ou ele pode promover o desenvolvimento de células T reguladoras.
Polimorfismos em Genes não HLA Associados à Autoimunidade
Análises de ligações de doenças autoimunes identificaram alguns genes associados à doença e muitas regiões cromossômicas onde se suspeitava da identidade dos genes associados, que ainda não estava estabelecida. A técnica de estudos de associação de abrangência genômica aumentou em muito a análise da base genética de doenças complexas, e atualmente conhecemos diversos genes que são associados a doenças autoimunes (Tabela 14-4). Antes que os genes que são mais obviamente validados sejam discutidos, é importante resumir algumas das características gerais destes genes.
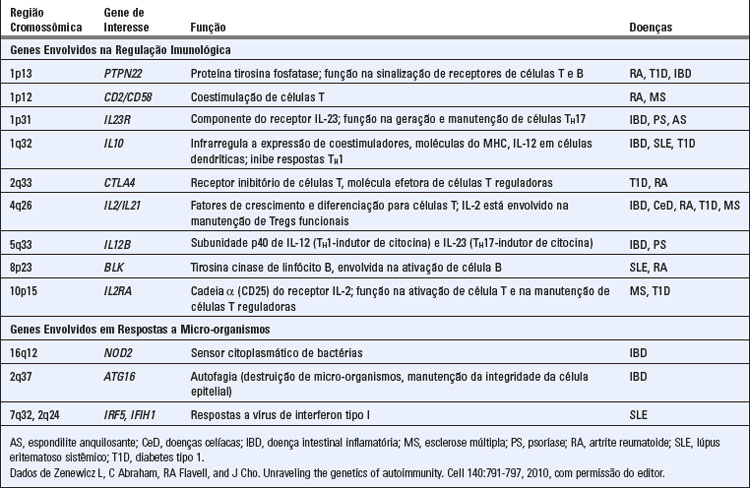
• Muitos dos polimorfismos associados a várias doenças autoimunes estão em genes que influenciam o desenvolvimento e regulação de respostas imunológicas. Embora esta conclusão pareça previsível, ela reforçou a utilidade das abordagens que são usadas para identificar genes associados a doenças.
• Polimorfismos diferentes podem proteger contra o desenvolvimento da doença ou aumentar a incidência da doença. Os métodos estatísticos usados para estudos de associação de abrangência genômica revelaram ambos os tipos de associações.
• Polimorfismos associados a doenças frequentemente são localizados em regiões não codificadoras dos genes. Este resultado inesperado sugere que os polimorfismos podem afetar a expressão das proteínas codificadas.
Alguns dos genes associados a doenças autoimunes humanas, que foram definidos por análise de ligações e por estudos de associação de abrangência genômica, são descritos brevemente a seguir.
• PTPN22. Uma variante da proteína tirosina fosfatase PTPN22 com ganho de função, que substitui uma arginina na posição 620 com um triptofano, é associado à artrite reumatoide, ao diabetes tipo 1, à tireoidite autoimune e a outras doenças autoimunes. Esta fosfatase ativada resulta na sinalização mais fraca do receptor de célula T e do receptor de célula B e pode contribuir assim para tolerância central ou periférica defeituosa nas células T e B. Um defeito parcial na tolerância em indivíduos com a variante triptofano poderia predispô-los à autoimunidade.
• NOD2. Polimorfismos nesse gene são associados com a doença de Crohn, um tipo de doença intestinal inflamatória. NOD2 é um sensor citoplasmático de peptidoglicanos da parede bacteriana (Cap. 4) e é expresso em múltiplos tipos de células, incluindo células epiteliais intestinais. Pensa-se que o polimorfismo associado à doença reduz a função do NOD2, que não pode oferecer defesa eficaz contra micro-organismos intestinais. Em consequência, esses micro-organismos conseguem atravessar o epitélio e iniciar uma reação inflamatória crônica na parede intestinal, que é a marca da doença intestinal inflamatória (Cap. 13).
• Insulina. Polimorfismos no gene da insulina que codificam números variáveis de sequência repetidas são associadas ao diabetes tipo 1. Estes polimorfismos podem afetar a expressão tímica da insulina. Postula-se que se a proteína é expressa em baixos níveis no timo devido a um polimorfismo genético, células T em desenvolvimento específicas para insulina podem não ser negativamente selecionadas. Estas células sobrevivem no repertório imunológico maduro e são capazes de atacar células β das ilhotas produtoras de insulina e causar o diabetes.
• CD25. Polimorfismos afetando a expressão de CD25, a cadeia α do receptor IL-2, são associados à esclerose múltipla, diabetes tipo 1, e outras doenças autoimunes. Não está claro se estas mudanças na expressão da CD25 afetam a manutenção de células T reguladoras ou a geração de células T efetoras e de memória induzida por IL-2; tanto defeitos na regulação como respostas efetoras e de memória excessivas podem contribuir para a autoimunidade.
• Receptor IL-23 (IL-23R). Alguns polimorfismos no receptor para IL-23 protegem contra o desenvolvimento de doença intestinal inflamatória e da doença cutânea psoríase. IL-23 é uma das citocinas envolvidas no desenvolvimento de células TH17 que disparam reações inflamatórias (Caps. 9 e 10). Pode ser que estes polimorfismos no IL-23R afetem as respostas de TH17 aos micro-organismos encontrados no trato intestinal e consequentemente o desenvolvimento de inflamação intestinal e cutânea.
• ATG16. Polimorfismos nesse gene também são associados a doença intestinal inflamatória. A proteína ATG16 é de uma família de proteínas envolvidas na autofagia, uma resposta celular à privação de nutrientes onde uma célula faminta “come” suas próprias organelas para fornecer substratos para a geração de energia e metabolismo. O processo de autofagia pode desempenhar uma função na manutenção de células epiteliais intestinais intactas ou a destruição de micro-organismos que entraram no citoplasma, mas não se sabe como o polimorfismo de ATG16 contribui para a doença intestinal inflamatória.
Houve um tremendo aumento no número de polimorfismos identificados em doenças inflamatórias, em grande parte devido a estudos de associação de abrangência genômica. Entretanto, estes estudos não identificam necessariamente um gene causal, mas podem indicar uma região onde um gene causal putativo está localizado. Estudos de associação de abrangência genômica não são adequados para a identificação de variantes raros que podem ser altamente penetrantes e podem, na verdade, ser a causa da doença. É provável que o advento do sequenciamento completo do genoma revele mais polimorfismos de nucleotídeos únicos (SNPs) em diversas doenças, então é certo que a lista aumente. Um dos grandes desafios no campo da genética de doenças complexas, incluindo doenças autoimunes e inflamatórias, é de correlacionar os polimorfismos genéticos com alterações fenotípicas. Até que isso aconteça, será difícil elucidar as funções destes genes na patogênese das doenças.
Anomalias em Genes Únicos que Causam Autoimunidade
Estudos com modelos de camundongos e pacientes identificaram vários genes que influenciam fortemente a manutenção da tolerância a antígenos próprios (Tabela 14-5). Ao contrário de polimorfismos em doenças complexas descritos anteriormente, estes defeitos em genes únicos são exemplos de distúrbios mendelianos nos quais a mutação é rara, mas tem alta penetrância, de modo que quase todos os indivíduos portadores da mutação são afetados. Muitos destes genes foram mencionados anteriormente nesse capítulo, quando discutimos os mecanismos de autotolerância. Apesar de estes genes estarem associados a doenças autoimunes raras, sua identificação fornece informações valiosas sobre a importância de várias vias moleculares na manutenção da autotolerância. Os genes conhecidos contribuem para os mecanismos de tolerância central (AIRE) estabelecidos, geração de células T reguladoras (FoxP3, IL-2, IL-2R), anergia e o funcionamento de células T reguladoras (CTLA-4), e deleção periférica de linfócitos T e B (Fas, FasL). Descrevemos aqui dois outros genes que são associados a doenças autoimunes em humanos.
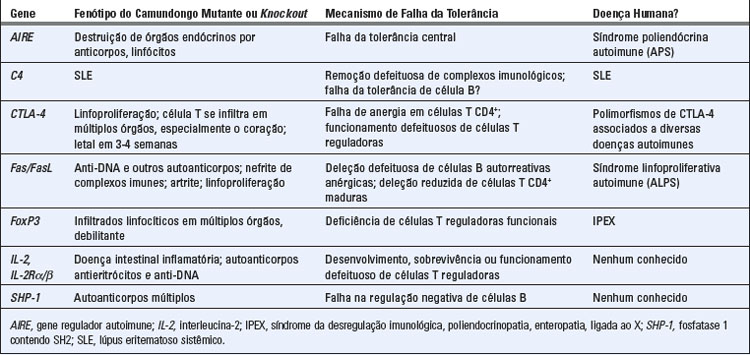
• Genes que codificam proteínas do complemento. Deficiências genéticas de várias proteínas do complemento, incluindo C1q, C2 e C4 (Cap. 12), são associadas a doenças autoimunes semelhantes ao lúpus. O mecanismo postulado para essa associação é que a ativação do complemento promove a remoção de complexos imunes circulantes e de corpos celulares apoptóticos, e, na ausência de proteínas complementares, estes complexos se acumulam no sangue e são depositados em tecidos e os antígenos de células mortas persistem.
• FcγRIIBPolimorfismos neste receptor inibitório Fc (Cap. 11) são associados a SLE em humanos, e a deleção genética deste receptor em camundongos resulta em uma doença autoimune semelhante ao lúpus. O mecanismo provável da doença é uma falha no controle de inibição de feedback de células B mediado por anticorpos.
Papel das Infecções na Autoimunidade
Infecções virais e bacterianas podem contribuir para o desenvolvimento e exacerbação da autoimunidade. Em pacientes e em alguns modelos animais, o início de doenças autoimunes é frequentemente associado a ou precedido por infecções. (Uma exceção notável e inexplicada é a linhagem de camundongos NOD [diabético não obeso], um modelo de diabetes tipo 1, no qual as infecções tendem a melhorar a insulite e o diabetes.) Na maioria destes casos, o micro-organismo infeccioso não está presente nas lesões e não é nem mesmo detectável no indivíduo quando a autoimunidade se desenvolve. Portanto, as lesões de autoimunidade não são devido ao próprio agente infeccioso, mas resultam de uma série de respostas imunológicas do hospedeiro que podem ser disparadas ou desreguladas pelo micro-organismo.
Infecções podem promover o desenvolvimento da autoimunidade por meio de dois mecanismos principais (Fig. 14-12).
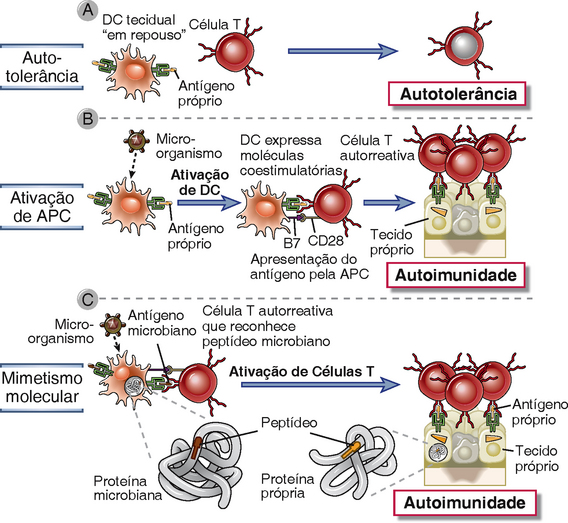
FIGURA 14-12 Papel das infecções no desenvolvimento da autoimunidade. A, Normalmente, o encontro de uma célula T autorreativa madura com um antígeno próprio apresentado por uma célula apresentadora de antígeno (APC) tecidual em repouso deficiente em coestimulador resulta na tolerância periférica por anergia. (Outros mecanismos possíveis de autotolerância não são mostrados.) B, Micro-organismos podem ativar as APC para expressar coestimuladores, e quando essas APC apresentam antígenos próprios, as células T autorreativas são ativadas em vez de ficar tolerantes. C, Alguns antígenos microbianos podem ter reações cruzadas com antígenos próprios (mimetismo molecular). Desta maneira, as respostas imunológicas iniciadas pelos micro-organismos podem ativar células T específicas para antígenos próprios.
• Infecções de tecidos específicos podem induzir respostas imunológicas naturais locais que recrutam leucócitos para dentro dos tecidos e resultam na ativação de APC teciduais. Estas APC começam a expressar coestimuladores e secretam citocinas ativadoras de célula T, resultando no colapso da tolerância da célula T. Assim, a infecção resulta na ativação de células T que não são específicas para o patógeno infeccioso; esse tipo de resposta é chamada de ativação espectadora. A importância da expressão aberrante de coestimuladores é sugerida por evidências experimentais de que a imunização de camundongos com antígenos próprios juntamente com adjuvantes fortes (que imitam micro-organismos) resulta no colapso da autotolerância e no desenvolvimento de doença autoimune. Em outros modelos experimentais, antígenos virais expressos em tecidos como células β de ilhotas induzem a tolerância de células T, mas a infecção sistêmica dos camundongos com o vírus resulta na falha da tolerância e na destruição autoimune das células produtoras de insulina.
• Micro-organismos também podem se envolver em receptores semelhantes a Toll (Toll-like receptors – TLR) em células dendríticas, levando à produção de citocinas ativadoras de linfócitos, e em células B autorreativas, levando à produção de autoanticorpos. Uma função da sinalização de TLR na autoimunidade foi demonstrada em modelos de SLE com camundongos.
• Micro-organismos infecciosos podem conter antígenos que têm reações cruzadas com antígenos próprios, então as respostas imunológicas aos micro-organismos podem resultar em reações contra antígenos próprios. Este fenômeno é chamado mimetismo molecular porque os antígenos do micro-organismo têm reações cruzadas com, ou imitam, antígenos próprios. Um exemplo de uma reação cruzada entre antígenos microbianos e próprios é a febre reumática, que se desenvolve depois de infecções estreptocócicas e é causada por anticorpos antiestreptocócicos que têm reações cruzadas com proteínas miocárdicas. Estes anticorpos são depositados no coração e causam a miocardite. O sequenciamento molecular revelou inúmeros trechos curtos de homologias entre proteínas miocárdicas e proteína estreptocócica.
Ainda falta estabelecer o significado de homologias limitadas entre antígenos microbianos e próprios em doenças autoimunes comuns, e tem sido difícil provar que uma proteína microbiana pode de fato causar uma doença que parece ser uma doença autoimune espontânea. Baseado em modelos de camundongos transgênicos, foi sugerido que o mimetismo molecular está envolvido no disparo da autoimunidade quando a frequência dos linfócitos autorreativos é baixa; nesta situação, o mimetismo microbiano do antígeno próprio serve para expandir o número de linfócitos autorrreativos acima de algum limiar patogênico. Quando a frequência de linfócitos autorreativos é alta, o papel dos micro-organismos pode ser o de induzir a inflamação tecidual, para recrutar linfócitos autorreativos para dentro do tecido, e para fornecer sinais secundários para a ativação destes linfócitos espectadores.
Algumas infecções podem proteger contra o desenvolvimento da autoimunidade. Estudos epidemiológicos sugerem que reduzir as infecções aumenta a incidência do diabetes tipo 1 e da esclerose múltipla, e estudos experimentais mostram que o diabetes em camundongos NOD é muito retardada se os camundongos forem infectados. Parece paradoxal que infecções podem ser o início da autoimunidade e também inibir doenças autoimunes. Não se sabe como elas podem reduzir a incidência de doenças autoimunes.
Outros Fatores na Autoimunidade
O desenvolvimento da autoimunidade está relacionado a vários fatores além dos genes de suscetibilidade e infecções.
• Alterações anatômicas em tecidos, causadas por inflamação (possivelmente secundárias a infecções), lesão ou trauma isquêmico, podem levar à exposição dos antígenos próprios que normalmente ficam ocultos do sistema imunológico. Estes antígenos sequestrados podem não ter induzido a autotolerância. Portanto, se antígenos próprios anteriormente ocultos são liberados, eles podem interagir com linfócitos imunocompetentes e induzir respostas imunológicas específicas. Exemplos de antígenos anatomicamente ocultos incluem proteínas intraoculares e esperma. Acredita-se que uveítes e orquites pós-traumáticas são causadas por respostas autoimunes contra antígenos próprios que são liberados de seus lugares normais por um trauma.
• Influências hormonais desempenham um papel em algumas doenças autoimunes. Muitas doenças autoimunes têm maior incidência em mulheres do que em homens. Por exemplo, SLE afeta as mulheres quase 10 vezes mais frequentemente do que os homens. A doença semelhante ao SLE em camundongos (NZB × NZW)F1 se desenvolve apenas em fêmeas e é retardada por tratamento com androgênios. Não se sabe se esta prevalência resulta da influência dos hormônios sexuais ou de outros fatores relacionados ao gênero.
Doenças autoimunes estão entre os problemas científicos e clínicos mais desafiadores da imunologia. O conhecimento atual de mecanismos patogênicos ainda está incompleto, de modo que as teorias e as hipóteses continuam a exceder os fatos. Espera-se que a aplicação de novos avanços tecnológicos e a melhor compreensão da autotolerância levará a respostas mais claras e definitivas aos enigmas da autoimunidade.
• A tolerância imunológica é a não responsividade a um antígeno induzida pela exposição de linfócitos específicos àquele antígeno. A tolerância a antígenos próprios é uma propriedade fundamental do sistema imunológico normal, e a falha da autotolerância leva a doenças autoimunes. Antígenos podem ser administrados de modo a induzir tolerância em vez de imunidade, e isso pode ser explorado para a prevenção e tratamento de rejeição a transplantes e de doenças autoimunes e alérgicas.
• A tolerância central é induzida nos órgãos linfoides primários (timo e medula óssea) quando linfócitos imaturos encontram antígenos próprios presentes nestes órgãos. A tolerância periférica ocorre quando linfócitos maduros reconhecem antígenos próprios em tecidos periféricos sob condições específicas.
• Em linfócitos T, a tolerância central (seleção negativa) ocorre quando timócitos imaturos com receptores de alta afinidade reconhecem esses antígenos no timo. Algumas células T imaturas que encontram antígenos próprios no timo morrem e outras se desenvolvem como linfócitos T reguladores FoxP3+, que funcionam para controlar respostas a antígenos próprios em tecidos periféricos.
• Diversos mecanismos respondem pela tolerância periférica nas células T maduras. Em células T CD4+, a anergia é induzida pelo reconhecimento de antígenos sem coestimulação adequada ou pelo envolvimento de receptores inibitórios como CTLA-4 e PD-1. Células T reguladoras inibem respostas imunológicas em parte pela produção de citocinas imunossupressoras. Células T que encontram antígenos próprios sem outros estímulos ou que são estimuladas repetidamente morrem por apoptose.
• Em linfócitos B, a tolerância central é induzida quando células B imaturas reconhecem antígenos próprios multivalentes na medula óssea. O resultado comum é a aquisição de uma nova especificidade, chamada de edição de receptor, ou morte apoptótica das células B imaturas. Células B maduras que reconhecem antígenos próprios na periferia na ausência de células T auxiliares podem se tornar anérgicas e finalmente morrer por apoptose ou ficar funcionalmente irresponsiva devido à ativação de receptores inibitórios.
• A autoimunidade resulta de uma falha da autotolerância. Reações autoimunes podem ser iniciadas por estímulos ambientais, como infecções, em indivíduos geneticamente suscetíveis.
• As doenças autoimunes, em sua maioria, são poligênicas, e numerosos genes de suscetibilidade contribuem para o desenvolvimento da doença. A maior contribuição é dos genes do MHC; acredita-se que outros genes influenciem a seleção ou regulação de linfócitos autorreativos.
• Infecções podem predispor à autoimunidade por vários mecanismos, incluindo a expressão melhorada de coestimuladores em tecidos e reações cruzadas entre antígenos microbianos e antígenos próprios. Algumas infecções podem proteger os indivíduos da autoimunidade, por mecanismos desconhecidos.
Tolerância Imunológica, Mecanismos Gerais
Baxter AG, Hodgkin PD. Activation rules: the two-signal theories of immune activation. Nature Reviews Immunology. 2002;2:439-446.
Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590-597.
Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301-305.
Mueller DL. Mechanisms maintaining peripheral tolerance. Nature Immunology. 2010;11:21-27.
Parish IA, Heath WR. Too dangerous to ignore: self-tolerance and the control of ignorant autoreactive T cells. Immunology Cell Biology. 2008;86:146-152.
Redmond WL, Sherman LA. Peripheral tolerance of CD8 T lymphocytes. Immunity. 2005;22:275-284.
Shlomchik MJ. Sites and stages of autoreactive B cell activation and regulation. Immunity. 2008;28:18-28.
Singh NJ, Schwartz RH. Primer: mechanisms of immunologic tolerance. Nature Clinical Practice Rheumatology. 2006;2:44-51.
Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annual Review of Immunology. 2003;21:685-711.
Von Boehmer H, Melchers F. Checkpoints in lymphocyte development and autoimmune disease. Nature Immunology. 2010;11:14-20.
Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nature Reviews Immunology. 2002;2:11-19.
Hogquist KA, Baldwin TA, Jameson SC. Central tolerance: learning self-control in the thymus. Nature Reviews Immunology. 2005;5:772-782.
Kyewski B, Klein L. A central role for central tolerance. Annual Review of Immunology. 2006;24:571-606.
Mathis D, Benoist C. Aire. Annual Review of Immunology. 2009;27:287-312.
Nemazee D. Receptor editing in lymphocyte development and central tolerance. Nature Reviews Immunology. 2006;6:728-740.
Anergia; Receptores Inibitórios
Bandyopadhyay S, Soto-Nieves S, Macian F. Transcriptional regulation of T cell tolerance. Seminars in Immunology. 2009;19:180-187.
Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunological Reviews. 2008;224:166-182.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annual Review of Immunology. 2008;26:677-704.
Mueller DL. E3 ubiquitin ligases as T cell anergy factors. Nature Immunology. 2004;5:883-890.
Schwartz RH. T cell anergy. Annual Review of Immunology. 2003;21:305-334.
Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annual Review of Immunology. 2006;24:65-97.
Wells AD. New insights into the molecular basis of T cell anergy: anergy factors, avoidance sensors, and epigenetic imprinting. Journal of Immunology. 2009;182:7331-7341.
Zheng Y, Zha Y, Gajewski TF. Molecular regulation of T-cell anergy. EMBO Reports. 2008;9:50-55.
Bidere N, Su HC, Lenardo MJ. Genetic disorders of programmed cell death in the immune system. Annual Review of Immunology. 2006;24:321-352.
Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:321-326.
Strasser A, Puthalakath H, O’Reilly LA, Bouillet P. What do we know about the mechanisms of elimination of autoreactive T and B cells and what challenges remain. Immunology and Cell Biology. 2008;86:57-66.
Campbell DJ, Koch MA. Phenotypic and functional specialization of FoxP3+ regulatory T cells. Nature Reviews Immunology. 2011;11:119-130.
Curotto MA, Lafaille JL. Natural and adaptive Foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30:626-635.
Feurer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nature Immunology. 2009;10:689-695.
Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor FoxP3. Nature Immunology. 2005;6:331-337.
Josefowicz SZ, Rudensky A. Control of regulatory T cell commitment and maintenance. Immunity. 2009;30:616-625.
Li MO, Flavell RA. TGF-β: a master of all T cell trades. Cell. 2008;134:392-404.
Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity. 2009;30:656-665.
Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nature Reviews Immunology. 2010;10:490-500.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775-787.
Sansom DM, Walker LS. The role of CD28 and CTLA-4 in regulatory T cell biology. Immunological Reviews. 2010;212:131-148.
Shevach EM. Mechanisms of Foxp3+ T regulatory cell–mediated suppression. Immunity. 2009;30:636-645.
Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nature Immunology. 2008;9:239-244.
Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nature Immunology. 2010;11:7-13.
Ziegler SF. FoxP3: of mice and men. Annual Review of Immunology. 2006;6:209-226.
Mecanismos de Autoimunidade: Genética
Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genetics. 2008;4:e1000024.
Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annual Review of Immunology. 2009;27:363-391.
Pascual V, Chaussubel D, Banchereau J. A genomic approach to human autoimmune diseases. Annual Review of Immunology. 2010;28:535-571.
Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature. 2005;435:584-589.
Xavier RJ, Rioux JD. Genome-wide association studies: a new window into immune-mediated diseases. Nature Reviews Immunology. 2008;8:631-643.
Zenewicz L, Abraham C, Flavell RA, Cho J. Unraveling the genetics of autoimmunity. Cell. 2010;140:791-797.
Mecanismos de Autoimunidade: Fatores Ambientais
Bach J-F. Infections and autoimmune diseases. Journal of Autoimmunity. 2005;25(Suppl 1):74-80.
Chervonsky A. Influence of microbial environment on autoimmunity. Nature Immunology. 2010;11:28-35.
Fourneau JM, Bach JM, van Endert PM, Bach JF. The elusive case for a role of mimicry in autoimmune diseases. Molecular Immunology. 2004;40:1095-1102.