CAPÍTULO 18 Distúrbios de Hipersensibilidade
A imunidade adaptativa apresenta-se como uma importante função de defesa contra infecções microbianas, mas as respostas imunológicas são também capazes de causar lesão tecidual e doença. Os distúrbios causados pela resposta imunológica são chamados de doenças de hipersensibilidade. Esse termo originou-se da definição clínica de imunidade como “sensibilidade”, que se baseia na observação de que um indivíduo exposto a um antígeno apresenta uma reação detectável ou é “sensível” a esse antígeno em contatos subsequentes. Normalmente, as respostas imunológicas erradicam os micro-organismos infecciosos sem consequências mais sérias ao tecido hospedeiro. No entanto, essas respostas algumas vezes são controladas de forma inadequada, direcionadas inapropriadamente para os tecidos do hospedeiro ou desencadeadas de micro-organismos comensais ou antígenos ambientais que, em geral, são inofensivos. Nessas situações, a resposta imunológica normalmente benéfica é a causa da doença.
Neste capítulo, será descrita a patogênese de diferentes tipos de doenças de hipersensibilidade, com ênfase aos mecanismos efetores que causam lesão tecidual. Por fim, conclui-se com uma breve consideração sobre o tratamento das doenças autoimunes e exemplos de doenças que ilustram os princípios mais importantes.
CAUSAS DE DOENÇAS DE HIPERSENSIBILIDADE
As respostas imunológicas contra antígenos de diferentes fontes podem ser causa subjacente de distúrbios de hipersensibilidade.
• Autoimunidade: A falha dos mecanismos normais de autotolerância resulta em reações contra células e tecidos próprios, o que é chamado de autoimunidade (Cap. 14). As doenças causadas pela autoimunidade são denominadas doenças autoimunes. Estima-se que as doenças autoimunes afetem cerca de 2% a 5% da população de países desenvolvidos, além disso, a incidência dessas doenças está aumentando. Muitas dessas doenças são comuns em indivíduos com faixa etária entre 20 e 40 anos. Elas são mais comuns no sexo feminino do que no masculino, por razões ainda não esclarecidas. As doenças autoimunes são crônicas e debilitantes e representam um enorme custo médico e econômico. No início do século XXI, muitos novos tratamentos foram desenvolvidos para essas doenças com base em princípios científicos; entre eles, encontram-se os avanços mais impressionantes da medicina. Os mecanismos de autoimunidade são descritos no Capítulo 14; neste capítulo, serão abordadas as várias doenças autoimunes para ilustrar como a autoimunidade pode causar uma doença.
• Reações contra micro-organismos: Respostas imunológicas contra antígenos microbianos podem causar doenças se as reações forem excessivas ou se os micro-organismos forem incomumente persistentes. A resposta mediada por células T contra micro-organismos persistentes pode originar uma inflamação grave, algumas vezes, com formação de granulomas; essa é a causa de lesão tecidual na tuberculose e algumas outras infecções crônicas. Se anticorpos são produzidos contra antígenos microbianos, os anticorpos podem se ligar aos antígenos produzindo complexos, que se depositam nos tecidos e desencadeiam inflamação. Raramente, anticorpos ou células T contra micro-organismos podem provocar uma reação cruzada com o tecido hospedeiro. Em algumas doenças que envolvem o trato intestinal, denominadas doença intestinal inflamatória, a resposta imunológica é dirigida diretamente contra bactérias comensais que normalmente residem no intestino e não causam nenhum dano. Algumas vezes, a resposta imunológica que causa a doença pode ser totalmente normal, mas, no processo de erradicação da doença, os tecidos hospedeiros são lesados. Na hepatite viral, o vírus que infecta as células hepáticas não é citopático, mas é reconhecido como estranho pelo sistema imunológico. Os linfócitos T citotóxicos (CTL) tentam eliminar as células infectadas, e essa resposta imunológica normal danifica as células hepáticas. Esse tipo de reação normal não é considerado hipersensibilidade.
• Reações contra antígenos ambientais: A maioria dos indivíduos saudáveis não reage contra substâncias ambientais comuns, em geral, inofensivos, mas cerca de 20% da população responde de forma anormal a uma ou mais dessas substâncias. Esses indivíduos produzem anticorpos imunoglobulina E (IgE) que causam doenças alérgicas (Cap. 19). Alguns indivíduos tornam-se sensíveis a antígenos ambientais e químicos, quando em contato com a pele, e desenvolvem reações mediadas por células T que desencadeiam inflamação mediada por citocinas, resultando em sensibilidade de contato.
Em todas essas condições, os mecanismos de lesão tecidual são os mesmos que normalmente apresentam a função de eliminar patógenos infecciosos. Esses mecanismos incluem resposta imunológica inata, anticorpos, linfócitos T, várias outras células efetoras e mediadores da inflamação. O problema nas doenças de hipersensibilidade é que a resposta é desencadeada e mantida de forma inadequada. Como o estímulo para essa resposta imunológica anormal é difícil ou impossível de ser eliminado (p. ex., autoantígenos, micro-organismos comensais e antígenos ambientais) e o sistema imunológico possui muitos mecanismos de retroalimentação positivos (mecanismos de amplificação), uma vez que a resposta imune patológica inicia, é difícil controlá-la ou interrompê-la. Por isso, essas doenças de hipersensibilidade tendem a se tornar crônicas e a progredir, tornando-se grandes desafios terapêuticos para a medicina clínica.
MECANISMOS E CLASSIFICAÇÃO DAS REAÇÕES DE HIPERSENSIBILIDADE
As doenças de hipersensibilidade são comumente classificadas de acordo com o tipo de resposta imunológica e o mecanismo efetor responsável pela lesão celular e tecidual (Tabela 18-1). Essa classificação foi originalmente desenvolvida por dois imunologistas britânicos, Philip Gell e Robin Coombs.
TABELA 18-1 Classificação das Doenças Autoimunes
| Tipo de Hipersensibilidade | Mecanismos Imunopatológicos | Mecanismos de Lesão Tecidual e Doença |
|---|---|---|
| Hipersensibilidade imediata: tipo I | Anticorpos IgE | Mastócitos e seus mediadores (aminas vasoativas, mediadores lipídicos, citocinas) |
| Mediada por anticorpos: tipo II | Anticorpos IgM e IgG contra antígenos da superfície celular ou da matriz extracelular | |
| Mediada por complexos imunes: tipo III | Complexos imunes de antígenos circulantes e anticorpos IgM e IgG | Recrutamento e ativação de leucócitos mediados por receptor Fc e complemento |
| Mediada por células T: tipo IV |
• A hipersensibilidade imediata (hipersensibilidade do tipo I), causada por anticorpos IgE específicos para antígenos ambientais e mastócitos, é o tipo de doença de hipersensibilidade mais prevalente e será descrita separadamente no Capítulo 19. As doenças de hipersensibilidade imediata, comumente denominadas alergias ou desordens atópicas, constituem-se o protótipo das doenças causadas pela ativação da subpopulação de células TH2 de linfócitos T auxiliares (helper), em que as células T estimulam a produção de anticorpos IgE e inflamação.
• Anticorpos IgG e IgM podem causar lesão tecidual por meio da ativação do sistema complemento, pelo recrutamento de células inflamatórias e por interferir nas funções celulares normais. Alguns desses anticorpos são específicos para antígenos de determinadas células ou da matriz extracelular, sendo encontrados ligados a essas células ou tecidos ou como anticorpos livres na circulação; as doenças induzidas por tais anticorpos são chamadas de distúrbios de hipersensibilidade do tipo II. Outros anticorpos podem formar complexos imunes na circulação, os quais são subsequentemente depositados nos tecidos, em particular, nos vasos sanguíneos, causando lesão. As doenças por complexos imunes também são denominadas distúrbios de hipersensibilidade do tipo III.
• A lesão tecidual pode ocorrer devido à indução de inflamação pelos linfócitos T ou diretamente pela morte das células-alvo; tais condições são chamadas de distúrbios de hipersensibilidade do tipo IV. Sabe-se agora que muitas doenças de hipersensibilidade são causadas pela ativação de subpopulações TH1 ou TH17 das células T auxiliares, secretando citocinas que promovem inflamação, e a lesão tecidual é causada pelo recrutamento de leucócitos, principalmente neutrófilos e macrófagos. As células T auxiliares também estimulam a produção de anticorpos que danificam os tecidos e induzem à inflamação. Os CTL também podem contribuir para lesão tecidual em algumas doenças.
Essa classificação é útil porque tipos diferentes de respostas imunes patológicas exibem diferentes padrões de lesão tecidual e podem variar de acordo com as especificidades do tecido. Como resultado, diferentes mecanismos imunológicos causam distúrbios com características clínicas e patológicas distintas. No entanto, as doenças imunológicas na situação clínica frequentemente são complexas e causadas por uma combinação das respostas imunológicas humoral e mediada por células, bem como múltiplos mecanismos efetores. Essa complexidade não é surpreendente, uma vez que um único antígeno normalmente pode estimular tanto a resposta imunológica humoral como a celular, na qual vários tipos de anticorpos e células T efetoras são produzidos. Como múltiplos mecanismos podem estar envolvidos e a inflamação, normalmente inflamação crônica, é o componente principal das manifestações patológicas e clínicas dessas doenças, elas são algumas vezes agrupadas como doenças inflamatórias imunomediadas. Considerar essas doenças em conjunto também possui algum valor clínico porque, como será visto mais adiante neste capítulo, muitas são tratadas com os mesmos agentes biológicos ou agentes biológicos relacionados. Na discussão a seguir, serão utilizadas descrições que identificam os mecanismos patogênicos em vez de designações numéricas, que são menos informativas para tipos de hipersensibilidade.
Após, será realizada uma discussão sobre doenças imunomediadas por anticorpos e por células T.
DOENÇAS CAUSADAS POR ANTICORPOS
As doenças mediadas por anticorpos são produzidas tanto por anticorpos que se ligam a antígenos em determinadas células ou em tecidos extracelulares como por complexos antígeno-anticorpo que se formam na circulação e são depositados nas paredes dos vasos sanguíneos (Fig. 18-1). Para provar que uma doença é causada por anticorpos, seria necessário demonstrar que as lesões podem ser induzidas em um animal saudável por transferência adotiva de imunoglobulinas purificadas do sangue ou tecidos afetados de indivíduos com a doença. Um experimento produzido pela natureza é ocasionalmente visto em crianças cujas mães sofrem de doenças mediadas por anticorpos. Essas crianças podem nascer com manifestações transitórias de tais doenças devido à passagem transplacentária dos anticorpos. No entanto, em situações clínicas, o diagnóstico das doenças causadas por anticorpos ou complexos imunes, em geral, baseia-se na evidenciação de anticorpos ou complexos imunes na circulação ou depositados em tecidos, bem como por similaridades clinicopatológicas com doenças experimentais comprovadamente mediadas por anticorpos por transferência adotiva.
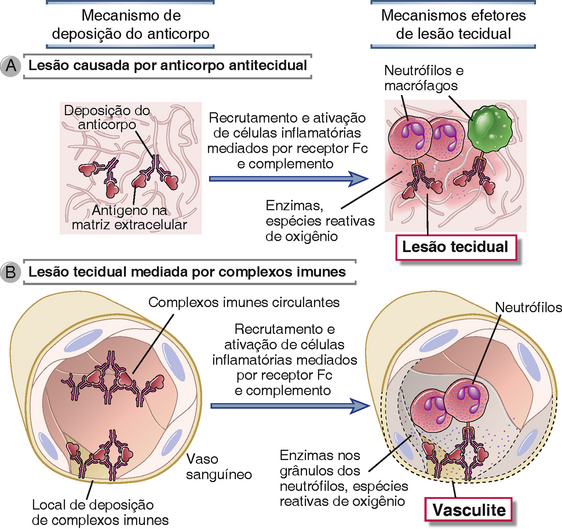
FIGURA 18-1 Tipos de doenças mediadas por anticorpos. Anticorpos podem se ligar especificamente a antígenos teciduais (A), ou podem se depositar como complexos imunes que são formados na circulação (B). Em ambos os casos, os anticorpos depositados induzem inflamação, desencadeando lesão tecidual.
Doenças Causadas por Anticorpos Contra Antígenos Celulares e Teciduais
Os anticorpos contra antígenos celulares ou da matriz causam doenças que afetam especificamente células ou tecidos onde esses antígenos estão presentes e, em geral, essas doenças não são sistêmicas. Os anticorpos contra antígenos teciduais causam doenças por meio de três mecanismos principais (Fig. 18-2).
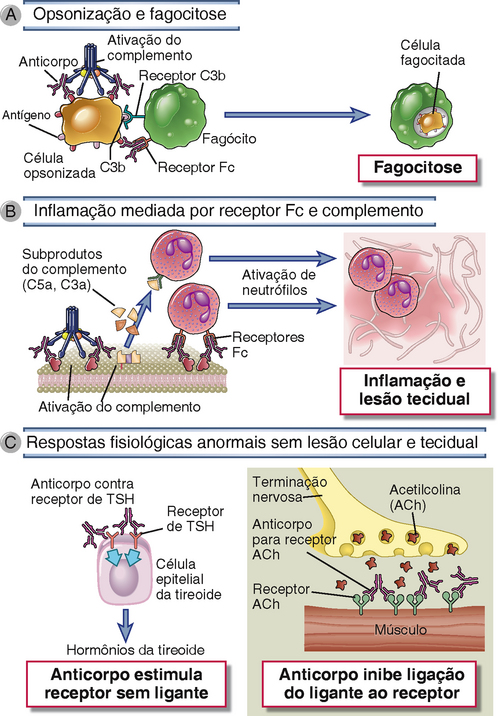
FIGURA 18-2 Mecanismos efetores de doença mediada por anticorpos. A, Anticorpos opsonizam células e podem ativar o complemento, gerando produtos do complemento que também opsonizam células, levando à fagocitose das células pelos receptores Fc ou receptores C3 dos fagócitos. B, Anticorpos recrutam leucócitos por ligação ao receptor Fc ou pela ativação do complemento e então há liberação de subprodutos que são quimiotáticos para leucócitos. C, Anticorpos específicos para receptores de superfície celular para hormônios ou neurotransmissores podem estimular a atividade dos receptores mesmo na ausência do hormônio (painel à esquerda) ou podem inibir ligando o neurotransmissor ao seu receptor (painel à direita). TSH, hormônio estimulante da tireoide.
• Os anticorpos que se ligam a antígenos de superfície celular podem opsonizar células diretamente ou podem ativar o sistema complemento, o que resulta na produção de proteínas do complemento que opsonizam células. As células opsonizadas são fagocitadas e destruídas por fagócitos que expressam receptores para porções Fc de anticorpos IgG e receptores para proteínas do complemento. Esse é o principal mecanismo de destruição celular na anemia hemolítica autoimune e na púrpura trombocitopênica autoimune. O mesmo mecanismo é responsável pela hemólise nas reações de transfusão (Cap. 16).
• Os anticorpos depositados nos tecidos recrutam neutrófilos e macrófagos, que se ligam a anticorpos ou se inserem a proteínas do complemento pelo Fc de IgG e receptores do complemento. Esses leucócitos são ativados pela sinalização de receptores, em particular, receptores Fc, e produtos de leucócitos, incluindo enzimas lisossomais e espécies reativas do oxigênio, são secretados e causam lesão tecidual. O mecanismo de lesão na glomerulonefrite mediada por anticorpos e em muitas outras doenças é inflamação e ativação de leucócitos.
• Os anticorpos que se ligam a receptores celulares normais ou outras proteínas podem interferir nas funções desses receptores ou proteínas e causar doença sem inflamação ou dano tecidual. As anormalidades funcionais mediadas por anticorpos são a causa da doença de Graves e da miastenia grave.
Os anticorpos que causam doenças celulares ou teciduais específicas geralmente são autoanticorpos produzidos como parte de uma reação autoimune contra antígenos nessas células ou tecidos. Exemplos desses autoanticorpos estão listados na Tabela 18-2. Menos comumente, os anticorpos podem ser produzidos contra um antígeno estranho (p. ex., micro-organismo) que é imunologicamente cruzado com um componente de tecidos próprios. Em uma rara consequência da infecção por estreptococos chamada de febre reumática, os anticorpos produzidos contra a bactéria reagem de forma cruzada com antígenos no coração, depositam-se no órgão e causam inflamação e lesão tecidual. Os depósitos teciduais de anticorpos podem ser detectados por exame morfológico em algumas dessas doenças, e a deposição do anticorpo, com frequência, está associada à ativação local do complemento, inflamação e lesão tecidual (Fig. 18-3A).
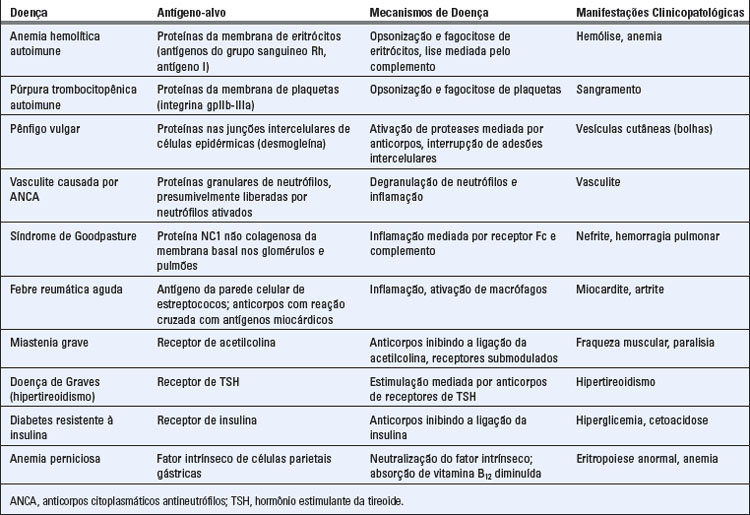
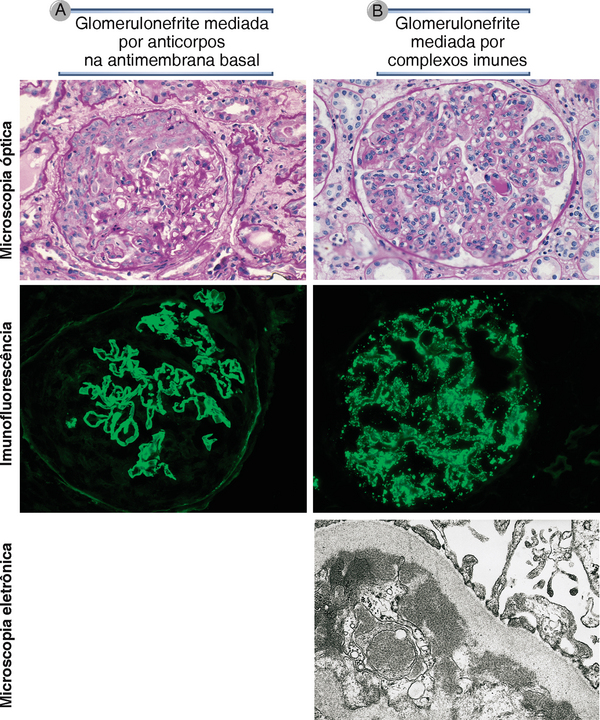
FIGURA 18-3 Características patológicas da glomerulonefrite mediada por anticorpos. A, Glomerulonefrite induzida por anticorpos contra a membrana basal glomerular (síndrome de Goodpasture): a microscopia óptica apresenta inflamação glomerular e graves danos, e a imunofluorescência exibe depósitos lineares de anticorpos ao longo da membrana basal. B, Glomerulonefrite induzida por deposição de complexos imunes (lúpus eritematoso sistêmico): a micrografia óptica apresenta inflamação neutrofílica, e a imunofluorescência e a microscopia eletrônica apresentam depósitos granulares de complexos de antígeno-anticorpo ao longo da membrana basal.
(Micrografias de imunofluorescência são cortesia de Dr. Jean Olson, Department of Pathology, University of California, San Francisco, e a micrografia eletrônica é cortesia de Dr. Helmut Rennke, Department of Pathology, Brigham and Women’s Hospital, Boston Massachusetts.)
Doenças Mediadas por Complexos Imunes
Os complexos imunes que causam doença podem ser compostos por anticorpos ligados tanto a antígenos próprios como a antígenos estranhos. As características patológicas das doenças causadas por complexos imunes refletem o local da deposição dos complexos imunes e não são determinadas pela fonte celular do antígeno. Assim, as doenças mediadas por complexos imunes tendem a afetar múltiplos tecidos e órgãos, embora alguns sejam particularmente suscetíveis, como rins e articulações.
Um astuto médico chamado Clemens von Pirquet suspeitou da ocorrência de doenças causadas por complexos imunes no início de 1911. Nessa época, a difteria era tratada com soro de cavalos imunizados com a toxina diftérica, o que é um exemplo de imunização passiva contra a toxina devido à transferência do soro contendo anticorpos antitoxina. Von Pirquet observou que a inflamação articular (artrite), rash e febre se desenvolviam em pacientes injetados com o soro de cavalo que continha a antitoxina. Duas características clínicas dessa reação sugerem que isso não se deve à infecção ou a componente da toxina do próprio soro. Primeiro, os sintomas apareciam mesmo após a injeção do soro do cavalo não contendo a antitoxina, de modo que as lesões não poderiam ser atribuídas aos anticorpos antidifteria. Segundo, os sintomas apareceriam, pelo menos, uma semana após a primeira injeção do soro de cavalo e mais rapidamente a cada injeção. Von Pirquet concluiu que essa doença devia-se à resposta do hospedeiro a algum componente do soro. Ele sugeriu que o hospedeiro produzia anticorpos para proteínas séricas do cavalo, e esses anticorpos formavam complexos com as proteínas injetadas, assim, a doença devia-se a anticorpos ou complexos imunes. Atualmente, sabe-se que suas conclusões estavam totalmente corretas. Ele denominou essa doença de doença do soro, que é o protótipo das desordens sistêmicas mediadas por complexos imunes.
Modelos Experimentais de Doenças Mediadas por Complexos Imunes
Doença do Soro
Muito do que se sabe atualmente sobre as doenças mediadas por complexos imunes baseia-se em análises de modelos experimentais da doença do soro. A imunização de um animal, como um coelho, com uma dose alta de antígeno proteico estranho desencadeia a formação de anticorpos contra o antígeno (Fig. 18-4). Esses anticorpos se ligam e formam complexos com anticorpos circulantes, que inicialmente são eliminados por macrófagos no fígado e baço. À medida que mais e mais complexos antígenos-anticorpos são formados, alguns deles são depositados no leito vascular. Nesses tecidos, os anticorpos dos complexos podem ativar o complemento, com uma queda concomitante dos níveis de complemento do soro. A ativação do complemento leva ao recrutamento e à ativação de células inflamatórias, com predominância de neutrófilos, nos locais de deposição dos complexos imunes, e os neutrófilos causam lesão tecidual. Os neutrófilos também se ligam a complexos imunes por meio dos receptores Fcγ, e a sinalização do receptor Fc ativa os leucócitos a produzir substâncias que lesam os tecidos, como nas doenças causadas por anticorpos contra tecidos fixos. Como os complexos são depositados principalmente em artérias pequenas, em glomérulos renais e na sinóvia das articulações, as manifestações clínicas e patológicas são vasculite, nefrite e artrite. Os sintomas clínicos, em geral, são de curta duração, e as lesões cicatrizam a menos que o antígeno seja injetado novamente. Esse tipo de doença é um exemplo de doença do soro aguda. Uma doença mais indolente e prolongada, chamada de doença do soro crônica, é produzida por múltiplas injeções de antígenos, que levam à formação de complexos menores depositados mais frequentemente nos rins, artérias e pulmões.
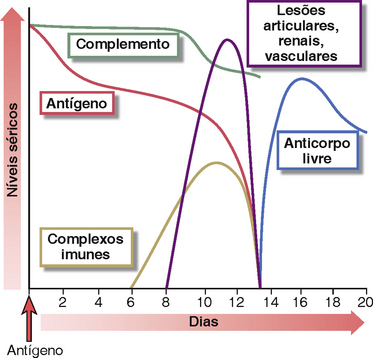
FIGURA 18-4 Sequências das respostas imunológicas na doença do soro aguda experimental. Injeção de albumina sérica bovina em um coelho desencadeia a produção de anticorpos específicos e a formação de complexos imunes. Esses complexos são depositados em múltiplos tecidos, ativam o complemento (levando à diminuição nos níveis séricos de complemento) e causam lesões inflamatórias, que determinam como os complexos e o antígeno remanescente são removidos e o anticorpo livre (não ligado ao antígeno) aparece na circulação.
(Adaptado de Cochrane CG. Immune complex-mediated tissue injury. In Cohen S, PA Ward, and RT McCluskey [Eds]. Mechanisms of Immunopathology. Werbel & Peck, New YorK, 1979, pp 29-48 ©1979, Wiley-Liss, Inc.)
Reação de Arthus
Uma forma localizada de vasculite experimental mediada por complexos imunes é chamada de reação de Arthus. Ela é induzida pela injeção de um antígeno subcutaneamente em um animal previamente imunizado ou em um animal que recebeu, via intravenosa, o anticorpo específico para o antígeno. Os anticorpos circulantes rapidamente se ligam ao antígeno injetado e formam complexos imunes depositados nas paredes das pequenas artérias do local em que ocorreu a injeção. Essa deposição origina uma vasculite cutânea local com necrose tecidual. Esse modelo tem sido utilizado para estudar as células e moléculas envolvidas nas doenças causadas por complexos imunes.
Patogênese de Doenças Mediadas por Complexos Imunes
Os complexos antígeno-anticorpo são produzidos durante as respostas imunológicas normais, porém eles causam doença somente quando produzidos em quantidades excessivas, não são eliminados de forma eficiente e ficam depositados nos tecidos. A quantidade de deposição de complexos imunes nos tecidos é determinada pela natureza dos complexos e as características dos vasos sanguíneos. Os complexos pequenos, muitas vezes, não são fagocitados e tendem a ser depositados nos vasos mais do que complexos grandes, que, em geral, são eliminados pelos fagócitos. Os complexos contendo antígenos catiônicos ligam-se avidamente aos componentes negativamente carregados da membrana basal dos vasos sanguíneos e glomérulos renais. Tais complexos normalmente produzem lesões teciduais graves e de longo prazo. Os capilares nos glomérulos renais e na sinóvia são vasos nos quais o plasma é ultrafiltrado (para formar urina e líquido sinovial, respectivamente) pela passagem através da parede dos capilares em alta pressão hidrostática; esses locais estão entre os mais comuns de deposição de complexos imunes. No entanto, os complexos imunes podem ser depositados em pequenos vasos de praticamente qualquer tecido. Os complexos imunes também podem se ligar a receptores Fc de mastócitos e leucócitos e ativar essas células para secretarem citocinas e mediadores vasoativos. Esses mediadores podem causar a deposição de mais complexos imunes nas paredes dos vasos por aumentarem a permeabilidade vascular e o fluxo sanguíneo.
A deposição de complexos imunes nas paredes dos vasos desencadeia uma inflamação mediada por receptores Fc e complemento, bem como lesão dos vasos e tecidos adjacentes. Os depósitos de anticorpos e complemento podem ser detectados nos vasos e, se o antígeno for conhecido, também é possível identificar moléculas dos antígenos nos depósitos (Fig. 18-3B).
Muitas doenças imunológicas sistêmicas em humanos são causadas pela deposição de complexos imunes nos vasos sanguíneos. Alguns exemplos comuns de doenças autoimunes por complexos imunes são o lúpus eritematoso sistêmico (SLE), no qual os complexos consistem em antígenos nucleares e anticorpos, e várias formas de nefrites e vasculites (Tabela 18-3). Em quase 50% dos casos de poliarterite nodosa, que é um tipo de vasculite mediada por complexos imunes envolvendo artérias musculares de tamanho médio, os complexos são compostos por antígenos virais e anticorpos, e a doença é uma complicação tardia de uma infecção viral, sendo o vírus da hepatite B o mais frequente. Esse também é o mecanismo de uma doença denominada glomerulonefrite pós-estreptocócica, que se desenvolve em raros casos após uma infecção estreptocócica e é causada por complexos de antígenos estreptocócicos e anticorpos depositados nos glomérulos renais.
TABELA 18-3 Exemplos de Doenças Humanas Mediada por Complexos Imunes
| Doença | Antígeno Envolvido | Manifestações Clinicopatológicas |
|---|---|---|
| Lúpus eritematoso sistêmico | DNA, nucleoproteínas, outros | Nefrite, artrite, vasculite |
| Poliarterite nodosa | Antígeno de superfície do vírus da hepatite B | Vasculite |
| Glomerulonefrite pós-estreptocócica | Antígenos da parede celular de estreptococos; pode ser “plantado” na membrana basal do glomérulo | Nefrite |
| Doença do soro | Proteínas variadas | Artrite, vasculite, nefrite |
DOENÇAS CAUSADAS POR LINFÓCITOS T
Os linfócitos T lesam tecidos tanto por desencadear inflamação como por eliminar diretamente células-alvo (Fig. 18-5). As reações inflamatórias são desencadeadas principalmente pelas subpopulações TH1 e TH17 das células T CD4+, as quais secretam citocinas que recrutam leucócitos. Em algumas desordens mediadas pelas células T, CTL CD8+ eliminam células-alvo que trazem antígenos associados ao complexo principal de histocompatibilidade (MHC) de classe I. As células T que causam lesão tecidual podem ser autorreativas ou podem ser específicas para antígenos proteicos estranhos que estão presentes em, ou ligados a, células e tecidos. A lesão tecidual mediada por linfócitos T também pode ser acompanhada de uma forte resposta imunológica contra micro-organismos persistentes, em especial, micro-organismos intracelulares que resistem à erradicação por fagócitos e anticorpos.
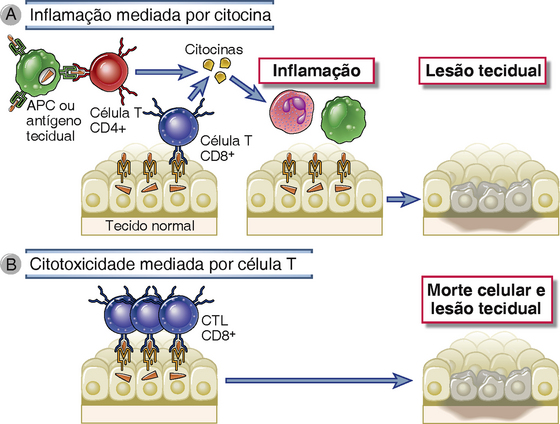
FIGURA 18-5 Mecanismo de doenças mediadas por células T. A, Em reações inflamatórias mediadas por citocinas, as células T CD4+ (e algumas vezes células T CD8+) respondem a antígenos teciduais por secretarem citocinas que estimulam inflamação e ativam fagócitos, levando à lesão tecidual. APC: célula apresentadora de antígeno. B, Em algumas doenças, os CTL CD8+ se direcionam diretamente para eliminar células teciduais.
Suspeita-se que o papel das células T como causa de doença imunológica específica está amplamente relacionado à demonstração de células T em lesões e o isolamento de células T específicas para antígenos próprios ou antígenos microbianos de diversos tecidos ou sangue de pacientes. Os modelos animais foram muito úteis para esclarecer a patogênese dessas desordens.
Doenças Causadas por Inflamação Mediada por Citocinas
Na inflamação imunomediada, as células TH1 e TH17 secretam citocinas que recrutam e ativam leucócitos. A IL-17, produzida pelas células TH17, promove o recrutamento de neutrófilos; o interferon γ (IFN-γ), produzido pelas células TH1, ativa macrófagos; e o fator de necrose tumoral (TNF) e as quimiocinas, produzidos pelos linfócitos T e outras células, estão envolvidos no recrutamento e na ativação de muitos tipos de leucócitos. A lesão tecidual é o resultado de produtos de neutrófilos e macrófagos ativados, como enzimas lisossomais, espécies reativas do oxigênio, óxido nítrico e citocinas pró-inflamatórias (Cap. 10). As células do endotélio vascular nas lesões podem expressar níveis aumentados de proteínas de superfície reguladas por citocinas, como moléculas de adesão e moléculas do MHC de classe II. A inflamação associada a doenças mediadas por células T normalmente é crônica, mas episódios de inflamação aguda podem se sobrepor em um histórico de inflamação crônica. A hipersensibilidade do tipo tardia (DTH) é um exemplo de tais reações inflamatórias e é descrita mais adiante. As reações inflamatórias crônicas, com frequência, produzem fibrose como resultado da secreção de citocinas e de fatores de crescimento pelos macrófagos.
Muitas doenças autoimunes específicas de órgãos são causadas pela interação de células T autorreativas com antígenos próprios, levando à liberação de citocinas e inflamação. Acredita-se que esse seja o mecanismo subjacente principal da artrite reumatoide, da esclerose múltipla, do diabetes tipo 1, da psoríase e de outras doenças autoimunes (Tabela 18-4). Algumas delas estão descritas com mais detalhes no final do capítulo.
TABELA 18-4 Doenças Mediadas por Células T
| Doença | Especificidade das Células T Patogênicas | Mecanismo Principal da Lesão Tecidual |
|---|---|---|
| Artrite reumatoide | ||
| Esclerose múltipla | Antígenos proteicos na mielina (p. ex., proteína básica da mielina) | |
| Diabetes melito tipo 1 | Antígenos de células β das ilhotas pancreáticas (insulina, ácido glutâmico descarboxilase, outros) | |
| Doença intestinal inflamatória | Inflamação mediada por citocinas TH1 e TH17 | |
| Miocardite autoimune | Proteína da cadeia pesada da miosina |
Exemplos de doenças humanas mediadas por células T estão listadas. Em muitos casos, a especificidade das células T e o mecanismo de lesão tecidual são inferidos com base na similaridade com modelos de animais experimentais das doenças.
As reações das células T contra micro-organismos e outros antígenos estranhos também podem levar à inflamação e lesão tecidual nesses locais de infecção ou de exposição do antígeno. As bactérias intracelulares, como o Mycobacterium tuberculosis, induzem uma forte resposta de células T e de macrófagos, o que resulta em inflamação granulomatosa e fibrose (descritas a seguir); a inflamação e a fibrose podem causar destruição tecidual excessiva e problemas funcionais, nesse caso, nos pulmões. A tuberculose é um bom exemplo de doença infecciosa na qual a lesão tecidual deve-se principalmente à resposta imunológica do hospedeiro (Cap. 15). As doenças cutâneas resultantes de exposição tópica a substâncias químicas e antígenos ambientais, denominadas sensibilidade de contato, devem-se a reações inflamatórias, presumivelmente desencadeadas por neoantígenos formados pela ligação das substâncias químicas a proteínas próprias. Tanto as células CD4+ como as CD8+ podem ser fonte de citocinas nas reações de sensibilidade de contato. Exemplos de sensibilidade de contato incluem o rash cutâneo por hera venenosa e carvalho venenoso (nos quais as células T reagem contra substâncias químicas, chamadas de urushiol, produzidas por essas plantas) e o eritema induzido pelo contato com várias substâncias químicas, como o tiuram, que é utilizado na confecção de luvas de látex. Algumas dessas reações tornam-se crônicas e clinicamente são chamadas de eczema. Acredita-se que as respostas das células T contra bactérias intestinais constituam a base de algumas formas de doenças intestinais inflamatórias.
A reação inflamatória clássica mediada pelas células T é chamada de hipersensibilidade do tipo tardia e é descrita a seguir.
Hipersensibilidade do Tipo Tardia
A hipersensibilidade do tipo tardia (DTH) é uma reação inflamatória lesiva mediada por citocinas que resultam da ativação de células T, em particular, células T CD4+. A reação é chamada de hipersensibilidade porque reflete uma resposta imunológica excessiva (reflexo da sensibilidade a um antígeno) e tardia porque normalmente se desenvolve após 24 ou 48 horas após o desafio do antígeno.
No modelo animal clássico de DTH, uma cobaia é primeiro imunizada pela administração de um antígeno de proteína em um adjuvante; essa etapa é chamada de sensibilização. Cerca de 2 semanas depois, o animal é desafiado subcutaneamente com o mesmo antígeno e a reação subsequente é analisada; essa etapa é chamada de fase de elicitação. Os humanos podem ser sensibilizados para reações de DTH por meio de infecções microbianas, por sensibilização de contato com substâncias químicas e antígenos ambientais ou por injeção subcutânea ou intradérmica de antígenos de proteínas (Fig. 18-6). A exposição subsequente ao mesmo antígeno (chamada de desafio) elicita a reação. Por exemplo, o derivado proteico purificado (PPD), um antígeno de proteína do Mycobacterium tuberculosis, elicita uma reação de DTH chamada de reação tuberculínica, quando é injetado em indivíduos que tenham sido expostos ao M. tuberculosis. O teste cutâneo tuberculínico positivo é amplamente utilizado como indicador clínico para evidência de infecção tuberculosa ativa ou prévia.
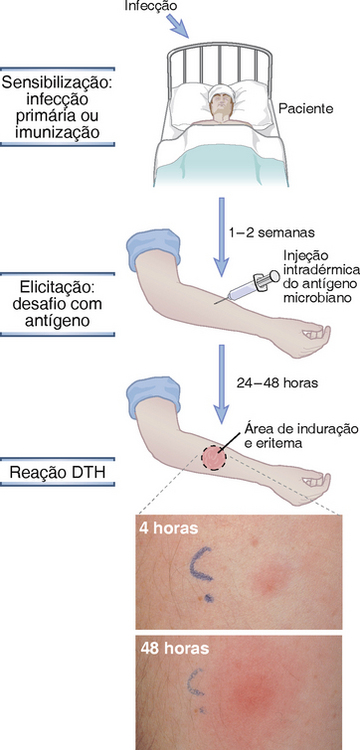
FIGURA 18-6 Reação de hipersensibilidade do tipo tardia. A infecção ou imunização (vacinação) sensibiliza um indivíduo e o desafio subsequente com um antígeno do agente infeccioso suscita uma reação de DTH. A reação manifesta-se por induração com vermelhidão e tumefação no local do desafio, que é indetectável em até 4 horas e apresenta um pico em 48 horas.
(Cortesia de Dr. J. Faix, Department of Pathology, Stanford University School of Medicine, Palo Alto, California.)
A resposta característica de DTH se desenvolve em 24 ou 48 horas. Cerca de 4 horas após a injeção do antígeno, neutrófilos acumulam-se ao redor de vênulas pós-capilares no local da injeção do antígeno. Por volta de 12 horas, o local da injeção do antígeno torna-se infiltrado por células T e monócitos sanguíneos, também organizados em uma distribuição perivenular (Fig. 18-7). As células endoteliais que recobrem essas vênulas tornam-se dilatadas mostram organelas biossintéticas aumentadas e há vazamento de macromoléculas plasmáticas. Há escape de fibrinogênio de vasos sanguíneos que circundam esses tecidos, onde ele é convertido em fibrina. A deposição de fibrina e, em menor grau, o acúmulo de células T e monócitos no espaço tecidual extravascular ao redor do local da injeção causam tumefação do tecido e ele torna-se firme (indurado). O endurecimento (induração), uma característica diagnóstica de DTH, é detectável cerca de 18 horas após a injeção do antígeno e é máxima por volta de 24 a 48 horas. Clinicamente, a perda da resposta de DTH a antígenos encontrados amplamente (p. ex., antígenos de Candida) é uma indicação de funcionamento deficiente de células T, uma condição conhecida como anergia. (Essa perda geral da responsividade imunológica é diferente da anergia linfocitária, um mecanismo para manter a tolerância a antígenos específicos, discutido no Cap. 14.)
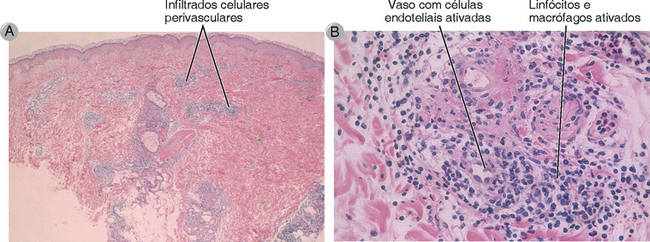
FIGURA 18-7 Morfologia de uma reação de DTH. A, O exame histopatológico da reação cutânea ilustrada na Figura 18-6 exibe infiltrados perivasculares de células mononucleares na derme. B, Em maior aumento, o infiltrado consiste em linfócitos ativados e macrófagos que circundam os vasos sanguíneos pequenos nos quais as células endoteliais também estão ativadas.
(Cortesia de Dr. J. Faix, Department of Pathology, Stanford University School of Medicine, Palo Alto, California.)
Embora a DTH tradicionalmente seja considerada um reação lesiva mediada por TH1, outras células T podem contribuir para a inflamação. Em algumas lesões por DTH, neutrófilos se sobressaem, sugerindo o envolvimento de células TH17. Em infecções por parasitas helmínticos, reações contra ovos de parasitas iniciam uma resposta DTH com forte componente eosinofílico. Nesses casos, um papel para citocinas TH2 foi demonstrado. As células T CD8+ também produzem IFN-γ e podem contribuir para algumas reações de DTH, em especial, as cutâneas.
As reações de DTH crônicas podem se desenvolver se a resposta de TH1 a uma infecção ativar macrófagos, mas falhar ao eliminar os micro-organismos fagocitados. Se os micro-organismos estão localizados em uma área pequena, a reação produz nódulos de tecido inflamatório chamados de granulomas (Fig. 18-8A). A DTH crônica, como exemplificado na inflamação granulomatosa, é causada pela sinalização prolongada das citocinas (Fig. 18-8B). Em tais reações, as células T ativadas e os macrófagos continuam a produzir citocinas e fatores de crescimento, que amplificam as reações de ambos os tipos de células e progressivamente modificam o ambiente tecidual. O resultado é um ciclo de lesão tecidual e inflamação crônica seguido pela reposição com tecido conjuntivo (fibrose). Nas reações de DTH crônicas, os macrófagos ativados também sofrem alterações em resposta a sinais de citocinas persistentes. Esses macrófagos desenvolvem o aumento de organelas citoplasmáticas e citoplasma e histologicamente podem lembrar células epiteliais cutâneas, por isso, são algumas vezes chamadas de células epitelioides. Os macrófagos ativados podem se fusionar e formar células gigantes multinucleadas. A inflamação granulomatosa é uma tentativa de conter uma infecção, mas também é causa significativa de lesão tecidual e de dano funcional. Esse tipo de inflamação é uma resposta característica a alguns micro-organismos resistentes, como o M. tuberculosis e alguns fungos, e representa uma forma de DTH crônica com fibrose. Grande parte da dificuldade respiratória associada à tuberculose ou infecção fúngica crônica pulmonar é causada pela substituição de tecido pulmonar normal por tecido fibrótico e não está diretamente relacionada aos micro-organismos.
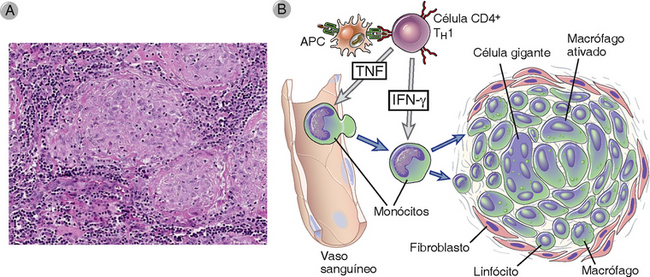
FIGURA 18-8 Inflamação granulomatosa. A, Linfonodo de um paciente com tuberculose contendo granulomas com macrófagos ativados, células gigantes multinucleadas e linfócitos. Em alguns granulomas, há uma área central de necrose. Estudos por imuno-histoquímica identificaram os linfócitos como células T. B, Mecanismos da formação do granuloma. As citocinas estão envolvidas na geração de células TH1, na ativação de macrófagos e no recrutamento de leucócitos. Reações prolongadas desse tipo desencadeiam a formação de granulomas.
Doenças Causadas por Linfócitos T Citotóxicos
A resposta por CTL a uma infecção viral pode desencadear lesão tecidual devido à morte de células infectadas, mesmo que o vírus em si não tenha efeito citopático. A principal função fisiológica dos CTL é eliminar micro-organismos intracelulares, principalmente vírus, eliminando as células infectadas. Alguns vírus lesam diretamente as células infectadas, então se diz que são citopáticos, enquanto outros não são. Como os CTL podem não ser capazes de distinguir entre vírus citopáticos e não citopáticos, eles eliminam as células infectadas por vírus, independentemente se a infecção por si é danosa ou não. Exemplos de infecções virais nas quais as lesões se devem à resposta por CTL do hospedeiro, e não ao vírus em si, incluem a coriomeningite linfocítica em camundongos e certas formas de hepatite viral em humanos (Cap. 15).
Os CTL podem contribuir para a lesão tecidual em distúrbios autoimunes causados principalmente por células T CD4+, como o diabetes tipo 1. São poucos os exemplos documentados de doenças autoimunes mediadas somente por CTL. A miocardite com infiltração cardíaca de células T CD8+ se desenvolve em camundongos e, algumas vezes, em humanos após a infecção pelo coxsackievirus B, resultando em uma forma de cardiomiopatia dilatada. Os pacientes e animais experimentais contêm CTL específicos para proteínas de miócitos. Postulou-se que as lesões cardíacas são iniciadas pela infecção viral e CTL vírus-específicos, e a lesão miocárdica leva à exposição ou alteração de antígenos próprios e ao subsequente desenvolvimento de CTLs autorreativos. No entanto, nessas formas de miocardite, as células T CD4+ e os anticorpos provavelmente também estão envolvidos na patogênese.
ABORDAGENS TERAPÊUTICAS PARA DOENÇAS AUTOIMUNES
Uma das mais impressionantes realizações da imunologia foi o desenvolvimento de novas terapias com base na compreensão da ciência básica e sua aplicação para doenças humanas. As terapias podem ser divididas em vários grupos.
Agentes Anti-inflamatórios
A base da terapia para doenças de hipersensibilidade, há muitos anos, têm sido os fármacos anti-inflamatórios, em particular, os corticosteroides. Tais fármacos são essenciais na redução da lesão tecidual, especificamente do componente inflamatório das respostas imunes patológicas.
Depleção de Células e Anticorpos
Acredita-se que os anticorpos monoclonais depletem todas as células linfoides, apenas células B ou apenas células T. Um desenvolvimento recente e um tanto surpreendente foi o uso bem-sucedido do anticorpo anti-CD20 (rituximabe), que depleta somente células B, para tratar doenças que se acreditava serem causadas, a princípio, por inflamação mediada por células T. Esse tratamento mostrou-se eficaz em alguns pacientes com artrite reumatoide e esclerose múltipla. A plasmaférese foi utilizada para eliminar autoanticorpos circulantes e complexos imunes.
Terapias Anticitocinas
Um grande número de citocinas envolvidas na inflamação é alvo de antagonistas específicos para o tratamento de doenças inflamatórias crônicas mediadas por células T (Tabela 18-5). O primeiro sucesso com essa classe de agentes biológicos veio com uma forma solúvel do receptor de TNF e anticorpos anti-TNF, os quais se ligam e neutralizam o TNF. Esses agentes são muito benéficos para muitos pacientes com artrite reumatoide, doença de Crohn e psoríase cutânea. Os antagonistas de outras citocinas pró-inflamatórias, como IL-1, a cadeia p40 que está presente na IL-12 e IL-23, IL-6 e IL-17A, e muitas outras, estão em uso ou em estudos clínicos para doenças inflamatórias.
TABELA 18-5 Exemplos de Antagonistas de Citocinas no Uso Clínico e em Estudos
| Citocina ou Receptor-alvo | Efeitos Biológicos Previstos do Antagonista | Indicações Clínicas |
|---|---|---|
| TNF | Inibe migração leucocitária para os locais de inflamação | Artrite reumatoide, psoríase, doença intestinal inflamatória |
| IL-1 | Inibe migração leucocitária para os locais de inflamação | Síndromes autoinflamatórias raras, gota grave, artrite reumatoide |
| IL-6 e receptor de IL-6 | Inibe síntese de proteínas de fase aguda, resposta de anticorpos? | Artrite idiopática juvenil, artrite reumatoide |
| IL-17 | Inibe recrutamento de leucócitos para os locais de inflamação | Artrite reumatoide, psoríase |
| Cadeia p40 da IL-12 e IL-23 | Inibe resposta TH1 e TH17 | Doença intestinal inflamatória, psoríase |
| Receptor de IL-2 (CD25) | Inibe proliferação de células T mediadas por IL-2 | Rejeição aguda do enxerto |
| IFN-α | Pode ter efeitos múltiplos na diferenciação TH1, produção de anticorpos | Lúpus eritematoso sistêmico |
| IL-4 | Inibe diferenciação TH2, produção de IgE | Asma |
| IL-5 | Inibe ativação de eosinófilos | Asma |
A TABLE lista exemplos de antagonistas contra citocinas (anticorpos ou receptores solúveis) que são aprovados para uso clínico ou estudos. IFN, interferon; IL, interleucina; TNF, fator de necrose tumoral.
Agentes que Inibem Interações Célula-célula nas Respostas Imunológicas
Os agentes que bloqueiam os coestimuladores B7 estão aprovados para tratamento da artrite reumatoide e psoríase, bem como têm sido testados no LES e outras doenças. Os anticorpos contra o ligante CD40 bloqueiam a ativação de células B e macrófagos mediada por células T, sendo benéficos para pacientes com doença intestinal inflamatória, mas um número pequeno de pacientes tratados desenvolveu episódios trombóticos, aparentemente porque essa molécula é expressa em plaquetas humanas (onde sua função é desconhecida). Os anticorpos contra integrinas foram utilizados para inibir a migração de leucócitos nos tecidos, em particular, no sistema nervoso central (SNC) na esclerose múltipla.
IgG Intravenosa
Altas doses de IgG intravenosa (IVIG) possuem efeitos benéficos em algumas doenças de hipersensibilidade. Não está claro como esse agente suprime a inflamação imunológica; uma possibilidade é que a IgG se ligue ao receptor Fc inibitório (FcγRIIB) em macrófagos e linfócitos B e então atenue a reposta inflamatória (Cap. 12). A IVIG também pode competir com anticorpos patogênicos pela ligação ao receptor Fc neonatal (FcRn), que tem como função em adultos proteger os anticorpos do catabolismo (Cap. 5), o que resulta em meia-vida reduzida de anticorpos patogênicos.
Existem tentativas de tratamentos mais específicos, como induzir tolerância nas células T produtoras de doença ou induzir células T regulatórias específicas para autoantígenos. A esclerose múltipla e o diabetes tipo 1 são duas doenças autoimunes nas quais os antígenos-alvo foram definidos; em ambos, estudos clínicos foram iniciados, nos quais os antígenos (peptídeos da proteína básica da mielina e insulina, respectivamente) serão administrados em pacientes de forma prevista a interromper respostas imunológicas específicas. Um risco de muitos tratamentos que bloqueiam vários componentes do sistema imunológico é que eles interfeririam no funcionamento normal do sistema imunológico para combate a micro-organismos e então tornariam os indivíduos suscetíveis a infecções. A tolerância a antígenos específicos evita esse problema visando seletivamente os linfócitos causadores de doença. Esses princípios gerais são semelhantes àqueles nos quais se baseia o tratamento de rejeição de transplante (Cap. 16).
DOENÇAS AUTOIMUNES ESPECÍFICAS: PATOGÊNESE E ESTRATÉGIAS TERAPÊUTICAS
Na seção seguinte, será descrita a patogênese de algumas doenças selecionadas, causadas por anticorpos e células T e a aplicação de novas terapias nessas doenças a fim de ilustrar princípios discutidos anteriormente.
Lúpus Eritematoso Sistêmico: o Protótipo da Doença Mediada por Complexos Imunes
O SLE é uma doença crônica autoimune multissistêmica, remitente e recidivante, que afeta predominantemente mulheres, com uma incidência de 1 em 700 entre mulheres com 20 e 60 anos (cerca de 1 em 250 entre mulheres negras) e em uma proporção de mulheres para homens de 10:1. As principais manifestações clínicas são erupções, artrite e glomerulonefrite, mas também são comuns anemia hemolítica, trombocitopenia e envolvimento do SNC. Muitos autoanticorpos diferentes são encontrados em pacientes com SLE. Os mais frequentes são os antinucleares, em particular, anticorpos anti-DNA; outros incluem anticorpos contra ribonucleoproteínas, histonas e antígenos nucleolares. Os complexos imunes formados desses autoanticorpos e seus antígenos específicos são responsáveis pela glomerulonefrite, artrite e vasculite que envolve as pequenas artérias por todo o corpo. A anemia hemolítica e a trombocitopenia se devem a autoanticorpos contra eritrócitos e plaquetas, respectivamente. O principal teste para diagnóstico dessas doenças é a presença de anticorpos antinucleares; os anticorpos contra a dupla fita de DNA nativo são específicos para SLE.
Patogênese do Lúpus Eritematoso Sistêmico
O LES é uma doença complexa na qual fatores genéticos e ambientais contribuem para a quebra da tolerância de linfócitos T e B autorreativos. Entre os fatores genéticos, estão, em particular, a herança de alelos HLA. O risco relativo para indivíduos com HLA-DR2 ou HLA-DR3 é de 2 a 3, e se ambos os haplótipos estiverem presentes, o risco relativo é de cerca de 5. As deficiências genéticas da via clássica do complemento, em especial, C1q, C2 ou C4, são observadas em cerca de 10% dos pacientes com SLE. As deficiências do complemento podem resultar em uma remoção defeituosa dos complexos imunes e das células apoptóticas, bem como falha da tolerância das células B. Um polimorfismo no receptor FcγRIIB inibitório de Fc foi descrito em alguns pacientes; isso pode contribuir para o controle inadequado da ativação das células B ou para a falha na atenuação da resposta inflamatória nas células imunes inatas. Muitos outros genes foram detectados por estudos genômicos, mas o papel de cada um não foi estabelecido, e sua contribuição no desenvolvimento da doença permanece desconhecida. Fatores ambientais incluem exposição à luz ultravioleta (UV). Postulou-se que isso desencadeia a morte de células por apoptose e liberação de antígenos nucleares.
Duas observações recentes são importantes para novas hipóteses em relação à patogênese do SLE. Primeiro, estudos com pacientes revelaram que células sanguíneas exibem uma “assinatura” molecular (padrão de expressão genético) impressionante que indica exposição ao TNF-α, um interferon tipo 1 produzido principalmente por células dendríticas plasmacitoides. Alguns estudos mostraram que estas células dendríticas, provenientes de pacientes com SLE, produzem, de forma anormal, grandes quantidades de IFN-α. Segundo, estudos em modelos animais mostraram que receptores semelhantes a Toll (Toll-like receptors – TLR), os quais reconhecem DNA e RNA, notavelmente o TLR9 reconhecedor de DNA, exercem um importante papel na ativação de células B específicas para antígenos nucleares próprios. Com base nesses estudos, um modelo para a patogênese do SLE foi proposto (Fig. 18-9). De acordo com esse modelo, a irradiação UV e outros fatores ambientais desencadeiam apoptose celular. A remoção inadequada do núcleo dessas células, em parte devido a defeitos nos mecanismos de remoção de proteínas do complemento e receptores, resulta em uma grande explosão de antígenos nucleares. Os polimorfismos em vários genes de suscetibilidade para o lúpus levam à capacidade defeituosa de manter a autotolerância de linfócitos B e T, razão pela qual linfócitos autorreativos permanecem funcionais. A falha da tolerância da célula B pode se dever à edição do receptor ou deleção defeituosa de células B imaturas na medula óssea ou tolerância periférica defeituosa. As células B autorreativas que não se apresentaram tolerantes são estimuladas pelos antígenos nucleares próprios, sendo produzidos anticorpos contra os antígenos. Os complexos de antígenos e anticorpos ligam-se a receptores Fc nas células dendríticas e no receptor de antígeno nas células B e podem ser internalizados. Os TLR engatam em componentes do ácido nucleico e estimulam células B a produzir autoanticorpos e a ativar células dendríticas, em particular, células dendríticas plasmacitoides, a produzir IFN-α, que potencializa a resposta imunológica e causa mais apoptose. Esse resultado em cadeia é um ciclo de liberação de antígeno e ativação imunológica que desencadeia a produção de autoanticorpos de alta afinidade.
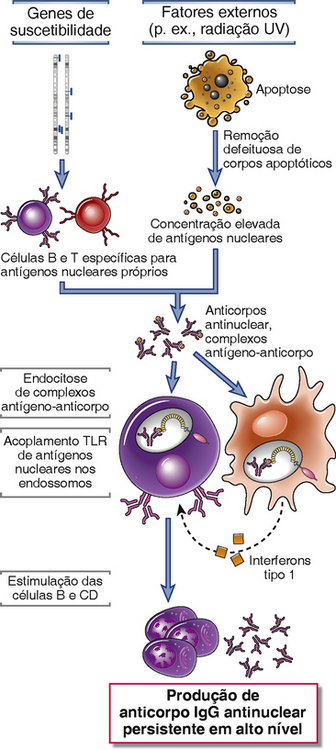
FIGURA 18-9 Um modelo para a patogênese do lúpus eritematoso sistêmico. Nesse modelo hipotético, vários genes de suscetibilidade interferem na manutenção da autotolerância, e fatores externos promovem a persistência de antígenos nucleares. O resultado é uma resposta de anticorpos contra antígenos nucleares próprios, que é amplificada pela ativação dependente de TLR das células dendríticas e células B por ácidos nucleicos e a produção de interferon tipo 1.
Novas Terapias para Lúpus Eritematoso Sistêmico
Os avanços recentes na compreensão do SLE estão contribuindo para novas abordagens terapêuticas. Estudos clínicos têm testado a eficácia de anticorpos anti-IFN-α na doença e tentativas de inibir sinais TLR têm sido consideradas. Há um grande interesse na depleção das células B pelo uso de um anticorpo contra a proteína CD20 de superfície da célula B. Um anticorpo que bloqueia o fator de crescimento BAFF da célula B foi recentemente aprovado para o tratamento do SLE.
Artrite Reumatoide
A artrite reumatoide (AR) é uma doença inflamatória que envolve pequenas e grandes articulações das extremidades, incluindo dedos, ombros, cotovelos, joelhos e tornozelos. A doença caracteriza-se pela inflamação da sinóvia associada à destruição da cartilagem articular e osso, apresentando um quadro morfológico indicativo de resposta imunológica local. As respostas imunológicas humoral e a mediada por células podem contribuir para o desenvolvimento da sinovite. Células CD4+ TH1 e TH17, linfócitos B ativados, plasmócitos e macrófagos, bem como outras células inflamatórias, são encontrados na sinóvia inflamada e, em casos graves, podem estar presentes folículos linfoides bem formados com centros germinativos. Inúmeras citocinas – incluindo IL-1, IL-8, TNF, IL-6, IL-17 e IFN-γ – têm sido detectadas no líquido sinovial (articular). Acredita-se que as citocinas recrutem leucócitos cujos produtos causam lesão tecidual e também ativam células sinoviais residentes a produzir enzimas proteolíticas, como colagenases, que medeiam a destruição de cartilagem, ligamentos e tendões das articulações. A atividade aumentada dos osteoclastos nas articulações contribui para a destruição óssea na AR, o que pode ser causado pela produção de citocinas RANK ligante (ativador do receptor do fator nuclear κβ) da família do TNF por células T ativadas. O ligante RANK liga-se a RANK, um membro da família de receptor TNF que é expresso em precursores do osteoclastos e induz sua diferenciação e ativação. As complicações sistêmicas da AR incluem vasculite, provavelmente causada por complexos imunes, e lesão pulmonar.
Embora muito da ênfase nos estudos de AR seja sobre o papel das células T, os anticorpos também podem contribuir para a destruição articular. As células B ativadas e plasmócitos estão frequentemente presentes na sinóvia das articulações afetadas. Os pacientes, com frequência, apresentam anticorpos circulantes para IgM e IgG, que reagem com as porções Fc (e raramente Fab) das suas próprias moléculas IgG. Esses anticorpos são chamados de fatores reumatoides e sua presença é utilizada como critério diagnóstico em testes para AR. Os fatores reumatoides podem participar da formação dos complexos imunes prejudiciais, mas seu papel patogênico ainda não foi estabelecido. Outro tipo de anticorpo detectado em pelo menos 70% dos pacientes é específico para peptídeos citrulinados cíclicos (CCP), que são derivados de certas proteínas modificadas em um ambiente inflamatório por conversão enzimática de resíduos da arginina para citrulina. Esses então chamados de anticorpos anti-CCP são marcadores diagnósticos para a doença e podem estar envolvidos na lesão tecidual.
Patogênese da Artrite Reumatoide
Assim como outras doenças autoimunes, a AR é um distúrbio complexo na qual fatores genéticos e ambientais contribuem para a quebra da tolerância imunológica para autoantígenos. A especificidade das células B e T patogênicas permanece desconhecida, por isso, a compreensão da sua patogênese está incompleta. A suscetibilidade para AR está ligada ao haplótipo HLA-DR4. Estudos recentes de associação de ligação e genômicos revelaram um grande número de polimorfismos genéticos associados à AR. Há uma associação com a codificação do gene da tirosina fosfatase, PTPN22, mas o papel dessa enzima na regulação dos linfócitos é pouco conhecido (Cap. 14).
A identificação de uma resposta imunológica anti-CCP levou a novas considerações em relação à patogênese da AR (Fig. 18-10). De acordo com um modelo, fatores ambientais, como tabagismo e algumas infecções, induzem a citrulinação de proteínas próprias, desencadeando a criação de novos epítopos antigênicos. Em indivíduos geneticamente suscetíveis, a tolerância a esses epítopos falha, o que resulta em uma resposta de células T e anticorpos contra as proteínas. Se essas autoproteínas modificadas também estão presentes nas articulações, as células T e os anticorpos atacam as articulações. As células TH17 e talvez TH1 secretam citocinas que recrutam leucócitos na articulação e ativam células sinoviais a produzir colagenases e outras enzimas. Assim, o resultado então é a destruição progressiva da cartilagem e do osso. A resposta imunológica na articulação pode ser forte o bastante para que tecidos linfoides terciários se formem na sinóvia e podem permanecer e propagar a reação inflamatória local.
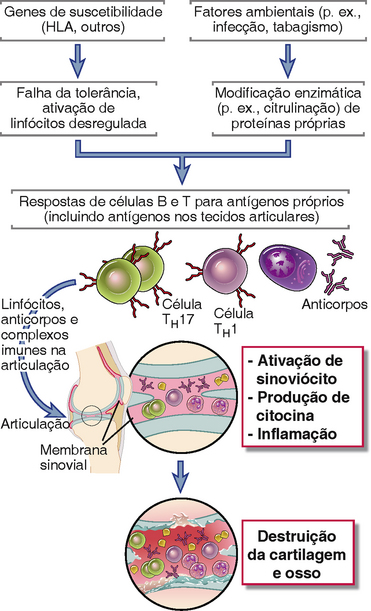
FIGURA 18-10 Um modelo para a patogênese da artrite reumatoide. De acordo com essa hipótese, proteínas citrulinadas induzidas por estímulos ambientais elicitam células T e uma resposta por anticorpos em indivíduos geneticamente suscetíveis. As células T e os anticorpos entram nas articulações, respondem a proteínas próprias e causam lesão tecidual principalmente por secreção de citocinas e talvez também por mecanismos efetores dependentes de anticorpos. As modificações das proteínas diferentes da citrulinação podem levar ao mesmo resultado.
Novas Terapias para Artrite Reumatoide
A compreensão do papel central das células T e citocinas na doença levou a um grande avanço no tratamento, no qual moléculas específicas passaram a ser visadas com base nessa compreensão científica. Entre essas novas terapias, as principais são os antagonistas contra o TNF, que têm alterado o curso da doença, em muitos pacientes, da inexorável destruição articular progressiva para uma latente, mas manejável, inflamação crônica. Um antagonista IL-1 e um anticorpo contra o receptor IL-6 foram aprovados para tratamento, como é uma proteína de fusão do domínio extracelular de CTLA-4 e a porção Fc de IgG, que se liga a moléculas B7 e bloqueia interações B7:CD28. Os anticorpos que bloqueiam IL-17 estão em estudos clínicos. O anticorpo anti-CD20 depletor de célula B traz benefícios para alguns pacientes. Os efeitos benéficos da depleção das células B não parecem ser atribuídos inteiramente à produção reduzida de autoanticorpos, o que sugere que as células B podem desempenhar outros papéis na doença, como apresentação de antígenos para células T patogênicas.
Esclerose Múltipla e Encefalomielite Autoimune Experimental
A esclerose múltipla (EM) é uma doença autoimune do SNC na qual células das subgrupos TH1 e TH17 das células T CD4+ reagem contra antígenos próprios da mielina, resultando em inflamação no SNC com ativação de macrófagos ao redor de nervos, cérebro e medula espinal, além de destruição da mielina, anormalidades na condução nervosa e defeitos neurológicos. Ela é a doença neurológica mais comum em adultos jovens. No exame patológico, existe uma inflamação na substância branca do SNC com desmielinização secundária. A esclerose múltipla é caracterizada clinicamente por fraqueza, paralisia e sintomas oculares com períodos de exacerbação e remissão; a imagem do SNC sugere que, nos pacientes com doença ativa, há frequente formação de novas lesões. A doença é modelada pela encefalomielite autoimune experimental (EAE) em camundongos, ratos, cobaias e primatas não humanos e é um dos modelos experimentais mais bem caracterizados de uma doença autoimune específica de um órgão mediada principalmente pelos linfócitos T. A EAE é induzida por imunização do animal com antígenos normalmente presentes na mielina do SNC, como a proteína básica da mielina, proteína proteolipídica e glicoproteína de oligodendrócito da mielina, junto a uma micobactéria inativada por calor como adjuvante, que é necessário para iniciar uma forte resposta por células T. Cerca de 1 ou 2 semanas após a imunização, os animais desenvolvem encefalomielite, caracterizada por infiltrado perivascular composto por linfócitos e macrófagos na substância branca do SNC, seguida de desmielinização. As lesões neurológicas podem ser leves e autolimitadas ou crônicas e recidivantes. Essas lesões resultam em uma paralisia progressiva ou remitente e recidivante. A doença também pode ser transferida para animais virgens (naïve) a partir de células T de animais doentes. Embora anticorpos contra antígenos de mielina tenham sido detectados em pacientes e em modelos de animais, o significado patogênico desses anticorpos não foi estabelecido.
Patogênese da Esclerose Múltipla
Existe uma abundância de evidências de que, em camundongos, a EAE é causada por células ativadas CD4+ TH1 e TH17 específicas para antígenos das proteínas da mielina. Por analogia com a doença experimental, acredita-se que a EM também seja causada por células TH1 e TH17 específicas para mielina e estas células têm sido detectadas em pacientes e isoladas no sangue e SNC. Como essas células são ativadas nos pacientes, ainda permanece um enigma. Tem sido sugerido que uma infecção, mais provavelmente uma infecção viral, ativa as células T autorreativas para mielina por meio do fenômeno de mimetismo molecular (Cap. 14). A autotolerância pode falhar em função da herança de genes de suscetibilidade. Gêmeos idênticos apresentam taxa de concordância de 25% a 40% para desenvolvimento de EM, enquanto gêmeos não idênticos apresentam uma taxa de concordância de 1%, o que indica fatores genéticos no desenvolvimento da doença. Os polimorfismos genéticos associados à EM incluem o lócus HLA, com HLA-DR2 sendo o elo mais forte. Estudos de associação genômicos têm revelado uma associação com polimorfismos na região não codificante do gene que codifica a cadeia α do receptor IL-2, CD25. A expressão do CD25 nas células T efetoras e de memória pode ser diferente em pacientes quando comparados com indivíduos saudáveis, mas como isso resulta em doença não foi esclarecido. Alguns estudos sugerem que células T regulatórias são defeituosas em pacientes com EM, mas como isso contribui para a falha da autotolerância também não se sabe. Uma vez que células T específicas para mielina são ativadas, elas migram para o SNC, onde encontram proteínas da mielina e liberam citocinas que recrutam e ativam macrófagos e mais células T, levando à destruição da mielina. Estudos sobre EAE sugerem que a doença é propagada por um processo denominado espalhamento de epítopo (Cap. 14). O tecido lesado resulta em liberação de novos antígenos proteicos e na expressão de novos epítopos anteriormente sequestrados que ativam mais células T autorreativas.
Novas Terapias para Esclerose Múltipla
A imunoterapia para EM baseou-se amplamente nas abordagens cujas bases científicas ainda não foram bem compreendidas. Isso inclui a administração de interferon-β, que pode alterar a resposta das citocinas, e o tratamento com polímeros randômicos de quatro aminoácidos, o qual é postulado para ligar moléculas HLA e bloquear a apresentação de antígenos. Mais recentemente, um anticorpo contra a integrina VLA-4 (Cap. 3) foi utilizado para bloquear a migração de leucócitos para dentro do SNC e mostrou-se benéfico para os pacientes. No entanto, em um pequeno número de pacientes, esse tratamento resultou em reativação de uma infecção latente pelo vírus JC, que causa uma doença grave e algumas vezes fatal do SNC. Outra droga recentemente aprovada para o tratamento de EM interfere na migração leucocitária. O fármaco, chamado de fingolimod (FTY720), bloqueia a via mediada pela esfingosina-1-fosfato de egresso da célula T dos tecidos linfoides (Cap. 3). Em um grande subconjunto de pacientes, a depleção de células B tem sido vista como de grande utilidade terapêutica. Sugere-se um importante papel das células B na ativação das células T patogênicas devido a esses resultados. Como a proteína básica da mielina é conhecida como um importante antígeno próprio que é alvo da resposta imunológica na EM, iniciaram-se tentativas de injetar peptídeos derivados desse antígeno nos pacientes, com esperança de se induzir tolerância ou de gerar células T regulatórias específicas para os antígenos relevantes.
Diabetes Melito Tipo 1
O diabetes melito tipo 1, antes denominado diabetes melito dependente de insulina, é uma doença metabólica multissistêmica resultante da insuficiência de produção insulina. Afeta cerca de 0,2% da população dos EUA, com um pico de idade por volta de 11 a 12 anos, sendo que a sua incidência está aumentando. A doença caracteriza-se por hiperglicemia e cetoacidose. As complicações crônicas do diabetes melito tipo 1 incluem aterosclerose progressiva das artérias, o que pode desencadear necrose isquêmica de membros e órgãos internos, e a obstrução microvascular, que causa danos à retina, a glomérulos renais e a nervos periféricos. Esses pacientes apresentam uma deficiência de insulina resultante da destruição imunomediada das células β produtoras de insulina das ilhotas de Langerhans no pâncreas, sendo necessária uma terapia com reposição contínua do hormônio.
Patogênese do Diabetes Tipo 1
Vários mecanismos podem contribuir para a destruição das células β, incluindo inflamação mediada por células CD4+ TH1 reativas a antígenos das ilhotas pancreáticas (incluindo insulina), lise das células das ilhotas mediada por CTL, produção local de citocinas (TNF e IL-1) que lesam as células das ilhotas e autoanticorpos contra células das ilhotas. Em raros casos nos quais as lesões pancreáticas foram examinadas no estágio mais inicial da doença, essas ilhas apresentaram necrose celular e infiltração linfocítica constituída de células T CD4+ e CD8+. Essa lesão é chamada de insulite. Os autoanticorpos contra as células das ilhotas e a insulina também são detectados no sangue desses pacientes. Em crianças suscetíveis que não desenvolveram diabetes ainda (como parentes dos pacientes), a presença de anticorpos contra as células das ilhotas é preditora do desenvolvimento de diabetes tipo 1, o que sugere que anticorpos anticélulas das ilhotas são patogênicos. Um modelo animal informativo sobre a doença é o camundongo diabético não obeso (NOD), que desenvolve diabetes espontaneamente. Nesse modelo, há evidência de sobrevida e função de células T regulatórias defeituosas, bem como resistência das células T efetoras para supressão.
Múltipos genes estão associados ao diabetes tipo 1. Grande atenção tem sido direcionada ao papel dos genes HLA. Entre 90% e 95% das pessoas caucasianas com diabetes tipo 1 apresentam HLA-DR3, ou DR4, ou ambos, em contraste com 40% dos indivíduos normais, e 40% a 50% dos pacientes são heterozigotos DR3/DR4, em contraste com 5% dos indivíduos normais. De forma interessante, atualmente, a suscetibilidade ao diabetes tipo 1 está associada a alelos DQ2 e DQ8 que, muitas vezes, sofrem um desequilíbrio de ligação com DR3 e DR4. Vários genes não HLA também contribuem para a doença. O primeiro a ser identificado foi o da insulina, que apresenta repetições em série na região promotora, estando associado à suscetibilidade à doença. O mecanismo dessa associação é desconhecido; ele pode estar relacionado ao nível de expressão da insulina no timo, o que determina se as células T insulina-específicas serão deletadas (selecionadas negativamente) durante a maturação. Vários outros polimorfismos foram identificados em pacientes e no camundongo NOD, incluindo os genes IL-2 e CD25. As consequências funcionais desses polimorfismos não são conhecidas. Alguns estudos sugerem que infecções virais (p. ex., com coxsackievirus B4) podem preceder o início do diabetes tipo 1, talvez por iniciar a lesão celular, induzindo inflamação e expressão de coestimuladores e desencadeando uma resposta autoimune. No entanto, dados epidemiológicos sugerem que infecções repetidas protegem contra diabetes tipo 1 e isso é similar no modelo NOD. De fato, tem sido postulado que uma das razões para a incidência aumentada de diabetes tipo 1 em países desenvolvidos é o controle de doenças infecciosas.
Novas Terapias para o Diabetes Tipo 1
As estratégias terapêuticas recentes mais interessante para o diabetes tipo 1 estão focadas em induzir tolerância com peptídeos diabetogênicos de antígenos de ilhotas pancreáticas (como a insulina) ou gerar ou fornecer células T regulatórias aos pacientes. Esses estudos clínicos ainda estão iniciando.
Doença Intestinal Inflamatória
A doença intestinal inflamatória consiste em duas desordens: a doença de Crohn e a colite ulcerativa, nas quais uma inflamação mediada por células T causa lesão intestinal. A doença de Crohn é caracterizada por inflamação crônica e destruição da parede intestinal, com frequente formação de fístulas. Na colite ulcerativa, as lesões são largamente confinadas à mucosa e consistem em úlceras com focos subjacentes de inflamação. A patogênese da doença intestinal inflamatória foi descrita no Capítulo 13. Novas terapias para essas doenças incluem anticorpos contra TNF, IL-17 e cadeia p40 de IL-12 e IL-23.
• Desordens causadas por respostas imunológicas anormais são chamadas de doenças de hipersensibilidade. As respostas imunológicas patológicas podem ser respostas autoimunes direcionadas contra antígenos próprios ou respostas descontroladas e excessivas para antígenos estranhos (p. ex., micro-organismos).
• Doenças de hipersensibilidade podem resultar de anticorpos que se ligam a células ou tecidos, complexos imunes circulantes depositados nos tecidos, ou linfócitos T reativos aos antígenos em tecidos.
• Os mecanismos efetores da lesão tecidual mediada por anticorpos são a ativação do complemento e a inflamação mediada pelo receptor Fc. Alguns anticorpos causam doença porque interferem no funcionamento celular normal sem produzir lesão tecidual.
• Os mecanismos efetores da lesão tecidual mediada por células T constituem-se nas reações inflamatórias induzidas por citocinas secretadas principalmente por células CD4+ TH1 e TH17 e lise celular por CTLs. A reação clássica mediada pela célula T é a hipersensibilidade do tipo tardia induzida pela ativação de células T previamente sensibilizadas e a produção de citocinas que recrutam e ativam vários leucócitos, principalmente macrófagos.
• O tratamento atual das doenças autoimunes tem como alvo a redução da resposta imunológica e as consequências lesivas da reação autoimune. A meta futura da terapia é inibir as respostas dos linfócitos específicos para autoantígenos e induzir tolerância a essas células.
• Doenças autoimunes como o lúpus eritematoso sistêmico, a artrite reumatoide, a esclerose múltipla e o diabetes tipo 1 ilustram muitos dos mecanismos efetores que causam lesão tecidual nas reações de hipersensibilidade e o papel dos genes de suscetibilidade e dos fatores ambientais no desenvolvimento da autoimunidade.
Davidson A, Diamond B. Autoimmune diseases. New England Journal of Medicine. 2001;345:340-350.
Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25-35.
Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619-630.
Anticorpos e Doenças Mediadas por Complexos Imunes
Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383-392.
D’Cruz DP, Khamashta MA, Hughes GR. Systemic lupus erythematosus. Lancet. 2007;369:587-596.
Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Advances in Immunology. 2006;92:1-69.
Jancar S, Sanchez Crespo M. Immune complex–mediated tissue injury: a multistep paradigm. Trends in Immunology. 2005;26:48-55.
Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nature Reviews Rheumatology. 2010;6:280-289.
Plotz PH. The autoantibody repertoire: searching for order. Nature Reviews Immunology. 2003;3:73-78.
Doenças Mediadas por Células T
Frohman EM, Racke MK, Raine CS. Multiple sclerosis—the plague and its pathogenesis. New England Journal of Medicine. 2006;354:942-955.
Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. Journal of Clinical Investigation. 2007;117:1119-1127.
Imboden JB. The immunopathogenesis of rheumatoid arthritis. Annual Review of Pathology. 2009;4:417-434.
Palmer MT, Weaver CT. Autoimmunity: increasing suspects in the CD4+ T cell lineage. Nature Immunology. 2010;11:36-40.